Neki neurološki poremećaji su rezultat patoloških efekata na nervni sistem tokom fetalnog razvoja i u ranom postnatalnom periodu. Takva stanja su statična, prvi klinički simptomi mogu se pojaviti tek u djetinjstvu, adolescencija ili kasnije.
Druga grupa bolesti nastaje kao rezultat mutacija specifičnih gena sa jasnim oblicima nasljeđivanja - dominantnim, recesivnim ili spolno povezanim. Ove bolesti se mogu otkriti pri rođenju, ali u mnogim slučajevima početak bolesti javlja se u djetinjstvu, adolescenciji ili odrasloj dobi. Ove bolesti su progresivne.
Treća grupa, koja se djelimično preklapa s drugom, uključuje progresivne degeneracije pojedinih odjela nervni sistem at genetska predispozicija, koji se realizuje pod uticajem faktora sredine.
Kongenitalne bolesti
Cerebralna paraliza
Definicija i etiologija
Motorni neurološki poremećaji, koji se uglavnom razvijaju kao posljedica pre- i perinatalne traume, ponekad su u kombinaciji s poteškoćama u učenju, poremećajima ponašanja i epilepsijom, ali nisu opasni po život. Faktori rizika za razvoj cerebralne paralize dati su u tabeli. 1, od kojih je najvažniji nedonoščad.
Tabela 1. Faktori rizika za cerebralnu paralizu (CP)
Istaknite raznih oblika bolesti.
- Spastična diplegija(Littleova bolest) je kongenitalna spastična parapareza, ponekad u kombinaciji sa skraćivanjem i deformacijom nogu, stvarajući probleme pri hodu. Gornji ekstremiteti su manje zahvaćeni, iako može doći do nespretnosti ruku.
- Spastična hemiplegija- čest oblik, obično povezan sa gubitkom vidnih polja (hemianopsija) i poremećenom osetljivošću na polovini tela (hemihipestezija), teškoćama u učenju i epilepsijom.
- Atetoidna cerebralna paraliza- koreoatetoidni pokreti koji se razvijaju u ranom djetinjstvu, kognitivne funkcije su obično normalne, teškoće u komunikaciji kod djece mogu biti posljedica dizartrije. Prethodno teški oblik ovog sindroma smatra se neonatalnom hiperbilirubinemijom ( kernicterus).
- Drugi oblici može biti teži (tetraplegija), manifestirati se kao izolirana ataksija ili imati kombinirane manifestacije.
Tretman
Liječenje cerebralne paralize uključuje hiruršku korekciju pridruženih skeletnih abnormalnosti i primjenu antikonvulziva za epilepsiju. Unatoč fizikalnoj terapiji, kontrakture i drugi deformiteti mogu zahtijevati ortopedsku intervenciju. Mnoga djeca zahtijevaju posebne obrazovne programe.
Spinalni disrafizam
Definicije
Kršenje normalnog zatvaranja neuralna cijev tokom embrionalnog razvoja, zajedno sa defektima kože i abnormalnim razvojem koštanih struktura, dovodi do disrafizam. Ako je zahvaćen mozak, takve ozljede mogu biti nespojive sa životom, kao u slučaju anencefalija kada nema mozga i lobanje.
Spinalni disrafizam zahvaća pretežno lumbosakralni region i može varirati po težini. At spinalna mijelomeningocela fragmenti kičmena moždina se izdaju u meningealnu kapsulu u lumbosakralnoj regiji. Obje anomalije su povezane s hidrocefalusom (vidi dolje). Skriveni rascjep luka pršljenova- većina lagana forma disrafizam, u kojem dolazi do nefuzije lukova kralježaka. Atipično locirani čuperak dlake u lumbalnoj regiji, lokalna depresija ili džep može se nalaziti na površini postojećeg defekta. Kod mnogih zdravih ljudi, okultna spinalna bifida je slučajan radiološki nalaz.
Etiologija
Uzroci okultne spinalne bifide nisu poznati. Postoje faktori rizika:
- defekt neuralne cijevi kod brata ili sestre
- trisomija 13. ili 18. hromozoma
- deficit folna kiselina on ranim fazama trudnoća
- blago povećanje rizika povezano je sa upotrebom antikonvulzanata tokom trudnoće, posebno natrijum valproata.
Tretman
Liječenje novorođenčadi s teškim razvojnim anomalijama je kompleksno hirurške operacije i predstavlja ozbiljan etički problem, budući da većina preživjele djece ostaje invalidna. Trenutno se dijagnoza može postaviti ultrazvučnim pregledom embrija, kao i na osnovu visoki nivo alfa-fetoprotein u majčinom serumu i amnionskoj tečnosti.
Latentna spinalna fuzija može se otkriti i kod odraslih osoba s rekurentnim bakterijskim meningitisom; nastaje zbog prisustva anastomoze između produbljivanja perkutane fistule i intratekalnog prostora. U isto vrijeme, pacijent može razviti sindrom cauda equina. MRI pregled (slika 1) može otkriti sindrom vezane vrpce, intraspinalni lipom ili krvarenje povezano s defektima kostiju. U ovoj situaciji hirurško lečenje može spriječiti napredovanje procesa.
Rice. 1. MRI kičmene moždine koja pokazuje da se kičmena moždina spušta u lumbalnu cisternu. Kičmena moždina sadrži šupljinu ispunjenu tekućinom, varijantu lumbalne siringomijelije (s). Zbog skolioze prikazane su dvije sagitalne fotografije
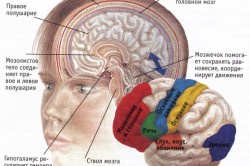
Infantilni hidrocefalus
Etiologija
Kongenitalni hidrocefalus je obično povezan s disrafizmom i sljedećim kongenitalnim anomalijama:
- stenoza Sylviusovog akvadukta
- Dandy-Walker sindrom- poteškoće u odljevu likvora iz četvrte komore s pridruženim razvojnim poremećajima vermisa malog mozga
- Arnold-Chiari malformacija- formiranje cerebralne kile i prolaps donjeg dijela moždanog stabla i malog mozga kroz foramen magnum.
Faktori kao što su traume, krvarenje, meningitis i, rjeđe, tumori mogu doprinijeti razvoju hidrocefalusa kod novorođenčadi.
Kliničke manifestacije
Moguće je da se veličina glave može povećati već u maternici, što može zakomplicirati proces porođaja. Najčešće novorođenčad izgleda normalno, ali kasnije se otkrije sljedeće:
- progresivno povećanje veličine glave (što je moguće u djetinjstvu, kada još nisu zarasli kranijalni šavovi), napetost fontanele, stanjivanje vlasišta s proširenim venama i fenomen „napuknutog lonca“ koji se otkriva perkusijom lubanje
- simptom "zalaska sunca" (poremećen pogled prema gore i nepotpuno zatvaranje očnih kapaka)
- zaostajati mentalni razvoj, poteškoće u učenju, epileptični napadi, atrofija optičkih nerava, spastična parapareza.
Tretman
Ako dijagnoza hidrocefalusa nije očigledna, može se potvrditi mjerenjem obima glave i rendgenskim pregledom ili, preciznije, CT skeniranjem lubanje. U teškim slučajevima neophodna je ventrikularna premosnica kako bi se spriječilo napredovanje bolesti. Priroda ostalih neurohirurških intervencija ovisi o karakteristikama patološkog procesa, na primjer, u slučaju Arnold-Chiari anomalije, vrši se dekompresija foramena magnuma. Kod neke djece s hidrocefalusom, proširenje ventrikula može se spontano povući, omogućavajući pacijentu da nastavi bez liječenja, ali može doći do ozbiljnih fizičkih i mentalnih deficita.
Anomalije strukture mozga
Opisane su brojne abnormalnosti razvoja mozga, od kojih mnoge mogu biti slučajni nalazi, npr. porencefalna cista povezani sa komorama, dok drugi mogu uzrokovati zastoj u razvoju, neurološke deficite i epileptički napadi.
Intrauterine infekcije
Uz široko uvođenje vakcinacije protiv rubeole i obaveznih skrining pregleda na rubeolu i sifilis kod trudnica, kongenitalne infekcije su rijetke.
Kongenitalna rubeola manifestira se bilateralnom kataraktom, gubitkom sluha, poteškoćama u učenju i urođenim srčanim manama.
Kongenitalni neurosifilis slično forma za odrasle, ali je karakterizirana brzim napredovanjem i specifičnim kliničkim manifestacijama - teškim gubitkom sluha, keratitisom i deformacijom zuba.
Genetski uslovljene bolesti nervnog sistema
Genetski defekti prvenstveno utiču na neuralnu cijev u razvoju, oštećujući razvoj specifičnih populacija neurona ili uzrokujući širenje efekta. S razvojem molekularne genetike, dijagnoza većine ovih stanja može se potvrditi analizom DNK
Cortex
Alchajmerova bolest, poput prionskih bolesti, može imati nasljedna predispozicija(vidi dolje).
Bazalni gangliji
Huntingtonova bolest
Autosomno dominantna bolest, u klasična verzija karakterizira progresivna koreja i demencija, počinje u dobi od 35-40 godina. Postoji dječji oblik u kojem rigidnost prevladava nad koreom (Vestfalska varijanta). Uprkos činjenici da je koreična hiperkineza zaustavljena terapija lijekovima, bolest je teško liječiti i smrt nastupa u roku od 15 godina. Morfološki pregled otkriva atrofiju kaudatnog jezgra uz generaliziranu cerebralnu atrofiju. Bolest se može blagovremeno dijagnosticirati pomoću DNK analize. To je dovelo do ozbiljnih etičkih problema zbog traumatskog utjecaja pozitivne dijagnoze na pacijenta i porodicu. Važno je razlikovati dijagnostička analiza za potvrdu sumnje na bolest i prediktivna analiza(identifikacija potencijalnog rizika kod potomstva). Pojedinačni zahtjev za preventivnu analizu se mora dogovoriti sa specijalistom.
Wilsonova bolest
Rijedak autosomno recesivni poremećaj metabolizma bakra. Koncentracije bakra u serumu i proteina za transport bakra ceruloplazmina u serumu su niske jer se bakar akumulira u tkivima, posebno u jetri i bazalnim ganglijama mozga. Prvi simptomi se mogu pojaviti u djetinjstvo zajedno sa cirozom jetre ili kod odraslih, kada prevladava neurološki poremećaji, kao što su akinetičko-rigidni sindrom, mišićna distonija, mentalni poremećaji, čak i psihoze. Bakar se takođe taloži u rožnjači ( Kaiser-Fleischer prsten, što se može otkriti pregledom prorezane lampe). Dijagnoza Wilsonove bolesti se zasniva na prisutnosti niskog serumskog bakra i ceruloplazmina, Kayser-Fleischer prstena, povećanom izlučivanju bakra u urinu i, ako je potrebno, biopsiji jetre. Adekvatna suportivna terapija dugi niz godina čuva život, pa čak i radnu sposobnost pacijenata, ali bez terapijske intervencije bolest je fatalna. Glavni lijek je penicilamin, koji vezuje bakar u helatni kompleks.
Mali mozak
Friedreichova ataksija
Rijetka recesivna bolest koja se manifestira progresivnom ataksijom, arefleksijom tetiva i patološkim refleksima stopala; javlja u detinjstvu. Uočavaju se i deformiteti skeleta: kifoskolioza i pes cavus, abnormalnosti na elektrokardiogramu, što ukazuje na kardiomiopatiju, koja je uzrok rane smrti. Trenutno je razvijena DNK dijagnostika.
Kasna ataksija
Nasljeđuje se autosomno dominantno sa očuvanim tetivnim refleksima (što se razlikuje od Friedreichove ataksije). Početak bolesti obično se javlja u odrasloj dobi, a tok cerebelarnih poremećaja je progresivan. Za neke spinocerebelarne ataksije (SCA), DNK dijagnoza je moguća. Upute za diferencijalnu dijagnozu cerebelarne ataksije date su u tabeli. 2.
Tabela 2. Diferencijalna dijagnoza cerebelarne ataksije
|
Nasljedni oblici Friedreichova ataksija Druge nasljedne ataksije Kongenitalne lezije Cerebelarna hipoplazija Dandy-Walker sindrom Arnold-Chiari malformacija Oblici traumatskih ozljeda Forms infektivnog porekla Cerebelarni apsces Tuberkuloza Postvirusno, na primjer nakon varičela Oblici upalne prirode Multipla skleroza Neoplazme Cerebelarni astrocitom, hemangioblastom, metastaze Paraneoplastični procesi Vaskularne lezije Hemorrhage miksedem (rijetko) Oblici uzrokovani toksičnim tvarima (uključujući lijekove) Alkohol Fenitoin Degenerativni oblici Višestruka sistemska atrofija |
Kortikospinalni trakt
Nasljedna spastična paraplegija
Karakterizira ga progresivna spastična parapareza, koja obično počinje u djetinjstvu, s tipičnim makazastim hodom i povezanim deformitetima skeleta. Nasljeđuje se autosomno dominantno. Nema poremećaja osjetljivosti ili disfunkcije sfinktera. Međutim, ima ih raznih složenih oblika kada spastična parapareza koegzistira s drugim neurološkim deficitom. Štaviše, javljaju se autosomno recesivni i spolno vezani načini nasljeđivanja.
Optički nerv
Leberova nasljedna optička neuropatija
Bolest se obično javlja u adolescenciji ili ranoj odrasloj dobi sa subakutnim jednostranim ili bilateralnim oštećenjem vida zbog oštećenja vidnog živca. Pacijenti obično imaju teške vidne deficite. Bolest je, iz nepoznatih razloga, češća među muškarcima, iako genetski defekt nije povezan sa spolom, već je mutacija mitohondrijalnu DNK(vidi dolje).
Ćelije prednjeg roga
Nasljedne spinalne amiotrofije
Zahvaćene su ćelije prednjih rogova; manifestira se kao mlitava paraliza i slabost u zahvaćenim mišićnim grupama. Postoji nekoliko varijanti bolesti, počevši od smrtonosnog dječjeg oblika ( Werdnig-Hoffmanova bolest) do blažeg generaliziranog oblika bolesti čiji se prvi simptomi javljaju u kasnom djetinjstvu ili adolescenciji ( Kugelberg-Welanderova bolest). Blaže varijante mogu biti ograničene na širenje jednog ekstremiteta ili druge vrste fokalnih lezija i nisu opasne po život. Tip nasljeđivanja je autosomno recesivno ili spolno vezano.
Periferni nervi
Charcot-Marie-Toothova bolest (CMT)
CMT je grupa klinički i genetski heterogenih poremećaja, a ne specifična bolest. Ova stanja se takođe opisuju kao nasledna motorna i senzorna neuropatija (HMSN).
Najčešća varijanta CMT1A nasljeđuje se autosomno dominantno i može se otkriti DNK dijagnostičkim metodama. Pacijenti imaju sporo progresivnu distalnu paralizu, koja u početku zahvaća mišiće anterolateralne noge. Ovo širenje u kombinaciji sa pes cavus izaziva karakterističan izgled donjih udova(Sl. 2). Tetivni refleksi su obično odsutni, a senzorni gubitak može biti relativno blag. Periferni živci su zadebljani i mogu se identificirati palpacijom. Elektroneurografija otkriva usporavanje širenja impulsa duž nerava. Histološki pregled perifernih nerava otkriva segmentnu demijelinizaciju, koja se manifestuje odgovarajućim promjenama EMG i ENMG, te pridruženom hipertrofijom. CMT2 je sličan tipu 1, ali se javlja češće kasno doba, brzina provodljivosti živaca ostaje relativno očuvana, što odražava preferencijalno oštećenje aksona, a ne demijelinizaciju.
Rice. 2.
Prognoza CMT-a je izuzetno varijabilna, čak i unutar iste porodice. Neki pacijenti se nađu vezani za njih invalidska kolica već u srednjim godinama, dok kod drugih bolest može biti asimptomatska.
Drugi, više rijetki uzroci periferna neuropatija može biti povezana sa specifičnim metaboličkim defektima, kao što su porodična amiloidoza, porfirija, leukodistrofija.
Mišići
Mišićne distrofije
Nasljeđuje se autosomno dominantno, autosomno recesivno ili spolno vezano.
Druge miopatije
Postoje podaci o brojnim drugim kongenitalne neuropatije, koji pogađaju uglavnom mišićno tkivo. Najzanimljivije mitohondrijski porazi. Glavni uzrok su mitohondrijalne mutacije, ne nuklearna DNK. Pacijenti imaju kroničnu progresivnu vanjsku oftalmoplegiju (obrazac donekle sličan očnom obliku mijastenije gravis) ili kombinaciju nekoliko drugih neuroloških i sistemskih manifestacija, kao što su ataksija, demencija, neuropatija, epilepsija, retinitis pigmentosa, generalizirana miopatija, kardiomiopatija i laktacidoza. Karakteristični prekršaji može se otkriti pomoću histološki pregled biopsije mišića ( "pokidana crvena vlakna"). Mutacije mitohondrijske DNK mogu se otkriti ispitivanjem krvi ili mišićnih ćelija. Tačkaste mutacije u mitohondrijskom genomu ukazuju na majčinski oblik nasljeđivanja u kojem potomstvo prima svu mitohondrijsku DNK iz jajeta.
Neurogenetski tumori
Mutacije gena sa pretpostavljenim funkcijama tumor supresori manifestiraju se kao tumori, hamartomi, ciste i druge neoplazme koje se razvijaju u raznih organa, ali sa pretežnom lokalizacijom u nervnom sistemu. Glavni simptomi ovih stanja, obično autosomno dominantnog tipa nasljeđivanja, dati su u tabeli. 3.
Tabela 3. Neurogenetski tumori
|
Bolest |
Nervni sistem |
Koža |
Ostali klinički simptomi |
|
Neurofibromatoza tip I (Recklinghausenova bolest) |
Periferni i spinalni neurofibromi Gliom optičkog živca Poteškoće u učenju |
Cafe-au-lait mrlje Dermatofibrom |
Lokalne promjene na šarenici Skeletni deformiteti Feohromocitom |
|
Neurofibromatoza tip II |
Bilateralni akustični neurom Meningioma Periferni i spinalni švanomi |
Nekoliko café au lait spotova |
Katarakta (obično asimptomatska) |
|
Tuberozna skleroza |
Problemi sa učenjem Epilepsija Tuberkuli i noduli mozga |
Adenoma lojne žlezde Subungualni fibromi Područja hipopigmentacije "šagrenska koža" |
Tumori retine Rabdomiom Ciste bubrega Angiolipomi bubrega |
|
Hipel-Lindauova bolest |
Cerebelarni hemangioblastom (moguća je i spinalna lokalizacija) |
|
Retinalni angiomi Ciste bubrega i drugih organa Karcinom bubrega Feohromocitom |
Pored gore navedenih, postoje i drugi neurokutani sindromi koji nisu povezani s tumorima i nisu nužno nasljedni. Na primjer, Sturge-Weberov sindrom se manifestira kao kombinacija cerebralne arteriovenske malformacije (sa kalcifikacijama) i rodni znak"boja porto vina" na istoj strani lica. Pacijenti obično imaju epilepsiju i hemiparezu kontralateralno u odnosu na malformaciju.
Neurodegeneracija
Jedna od najčešćih bolesti je Parkinsonova bolest. Ostalo patološka stanja, sa jasnom genetskom etiologijom, opisani su gore. U nastavku će biti opisane dvije neuronske populacije centralnog nervnog sistema koje su najosjetljivije na degenerativne bolesti:
- cerebralni korteks, posebno neuroni uključeni u svjesnu aktivnost
- motornih neurona (CMN i PMN).
demencija
Demencija se definira kao značajno oštećenje dvije ili više kognitivnih funkcija, od kojih je jedna memorija, dovoljno da ograniči izvođenje uobičajenih profesionalnih i društvene funkcije. Ovo stanje ne bi trebalo biti povezano sa delirijumom. Većina ljudi s demencijom ima degenerativno oštećenje mozga, iako su mogući i drugi uzroci (vidi dolje).
Alchajmerova bolest (AD)
AD je najčešći uzrok demencije i češći je kod starijih ljudi. Karakterizira ga stvaranje unutarćelijskih inkluzija u obliku neurofibrilarni zapleti, koji se sastoji od “uparenih spiralnih vlakana” i ekstracelularni plakovi koji sadrže amiloid, zajedno sa smrću neurona (slika 3).
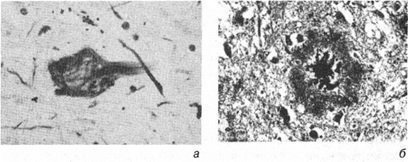
Rice. 3. Alchajmerova bolest (AD) - patološki kriterijumi, a - neurofibrilarni čvorići; b - neuritski plakovi
Etiologija i patogeneza
Hemijska analiza sadržaja plaka omogućila je da se utvrdi da je njegova glavna komponenta amiloid beta protein- fragment većeg molekula amiloidni prekursor protein (APP), kodiran genom hromozoma 21. Uloga amiloidnog proteina u patogenezi AD identificirana je u rijetkom porodičnom obliku uzrokovanom mutacijom ALD gena. Studija pacijenata s Downovim sindromom (trisomija 21) također je potvrdila patogenetsku ulogu amiloidnog proteina, s obzirom na činjenicu da pacijenti razvijaju rane simptome AD i da su u opasnosti od razvoja amiloidnih naslaga zbog prisustva dodatne kopije ALD gena. . Međutim, uzroci AD su mnogo složeniji od taloženja amiloida, budući da većina slučajeva nije porodična, au nekim porodične slučajeve Identifikovane su mutacije u genima koji nisu povezani sa ALD kodiranjem. Specifična izoforma proteina za transport lipida apolipoprotein E smatra se nezavisnim faktorom u nastanku i porodičnih i sporadičnih oblika astme.
Bez obzira na molekularne mehanizme koji leže u osnovi AD, patološki proces rezultira smrću neurona u područjima moždane kore povezanih s kognitivnim funkcijama, posebno u hipokampusu i susjednim strukturama, kao i u temporalni režanj neokorteks. Neke duboke strukture, kao što je bazalno jezgro Meynertovog frontalnog režnja, također su uključene u patološki proces. Tipično je oštećenje kolinergičkih neurona, što zahtijeva upotrebu lijekova koji poboljšavaju metabolizam acetilholina i pomažu poboljšanju pamćenja.
Kliničke manifestacije
U ranim stadijumima bolesti dolazi do smanjenja pamćenja, posebno kratkoročnog. Pacijenti imaju poteškoća u učenju i pamćenju nove informacije. Anamneza se često mora prikupljati od najbližih i rođaka, jer pacijent možda nije svjestan svojih problema.
Pojavljuju se daljnji poremećaji pamćenja i pažnje, koji dovode do dezorijentacije u vremenu, otežanog pronalaženja riječi i gubitka općeg znanja. Nedostaci percepcije mogu biti povezani sa halucinacijama i iluzijama. Konačno, dolazi do ozbiljnog globalnog gubitka kognitivnih funkcija – amnezije, afazije i agnozije. Ličnost se uništava poremećajima ponašanja, urinarnom i fekalnom inkontinencijom, povećavajući zavisnost od drugih u Svakodnevni život i smrt u roku od 5-10 godina.
Dijagnostika
Ne postoje specifični testovi za intravitalnu dijagnozu astme. Međutim, pažljiva primjena kliničkih dijagnostički kriterijumi daje tačan rezultat u 80% slučajeva. Vrlo je važno isključiti druge uzroke demencije, prvenstveno potencijalno izlječive (Tabela 4).
Tabela 4. Uzroci demencije
|
Nasljedni faktori Porodični oblici Alchajmerove bolesti Huntingtonova bolest Neke cerebelarne ataksije Wilsonova bolest Povrede Subduralni hematom Drugi oblici teških traumatskih ozljeda mozga Infekcije Subakutni sklerozirajući panencefalitis Demencija povezana sa AIDS-om Progresivna multifokalna leukoencefalopatija Cerebralni oblik Whippleove bolesti (zajedno s artritisom i crijevnim poremećajima) Multipla skleroza Vaskulitis, lupus, sarkoidoza Neoplazme Tumori frontalni režnjevi Višestruke metastaze na mozgu Hidrocefalus zbog stražnjeg tumora lobanjske jame(Pažnja! Hidrocefalus normalnog pritiska, čak iu odsustvu strukturnih lezija mozga, može izazvati demenciju; vidi tekst) Paraneoplastični procesi Vaskularne bolesti Multiinfarktna demencija Metabolički poremećaji miksedem Nedostatak vitamina B12 Hronična insuficijencija organa Opijenost Barbiturati, alkohol, olovo Neurodegeneracija Pickova bolest Prionske bolesti (urođene i stečene) Parkinsonova bolest i druge akinetičko-rigidne bolesti sindromi (vidi tekst) |
Tretman
Sistemske bolesti, poput infekcija, mogu zakomplikovati tok demencije, pa na njih treba obratiti pažnju opšte stanje pacijenta, uključujući izbjegavanje upotrebe sedativa (osim ako nije jasno naznačeno), pijenje alkohola i preopterećenost.
Jednostavan trening pamćenja, vođenje dnevnika i korištenje prečica i podsjetnika mogu biti od pomoći u ranim fazama bolesti. O bolesti treba obavijestiti i ured za vozačke dozvole.
Danas ne postoje lijekovi koji mogu radikalno promijeniti tok astme. Za poboljšanje pamćenja u ranim stadijumima bolesti koriste se različiti lijekovi koji aktiviraju kolinergičku transmisiju, čija je efikasnost maksimalna u prvim mjesecima upotrebe. Najčešće korišćeni inhibitori holinesteraze su donepezil, rivastigmin i galantamin (potonji se takođe koristi kao agonist nikotinskih receptora). Memantin inhibira glutamatergični prijenos i licenciran je u UK za liječenje umjerene do teške AD. Simptomatsko liječenje liječenje, kao što je donezepil, treba nastaviti samo ako pacijent ili njegovatelji prijave poboljšanje kvalitete života kao rezultat liječenja. Za korekciju emocionalni poremećaji Za astmu se koriste antidepresivi, antipsihotici i anksiolitici.
Za više kasne faze bolesti koje karakterizira sve veća ovisnost pacijenta o vanjskoj pomoći; glavni teret njege pada na bliske srodnike, često i starije osobe. Postoje ambulantne službe, kao što su grupne psihijatrijsku njegu, dnevne bolnice, organizacije za njegu i specijalizirane informativne agencije kao što je Alchajmerovo društvo.
Ostali uzroci demencije
Degenerativne bolesti
Prionske bolesti- grupa retkih neurodegenerativnih poremećaja kod životinja i ljudi, prethodno kombinovanih u jednu bolest zasnovanu na sličnim histološkim obrascima ( "spužvasti encefalitis"). Jedna od prvih opisanih bolesti ove grupe je Creutzfeldt-Jakobova bolest (CJD), koja je jedno od onih stanja koje se potencijalno može naslijediti i prenijeti putem zaraženog materijala.
Molekularna osnova bolesti je infektivni patogen - "prion". Posebnost ovog mikroorganizma je da se sastoji isključivo od proteina ( prionski protein, PrP) i vrlo je otporan na temperaturu i formaldehid. U neinficiranim ćelijama prisutna je izoforma PgR, kodirana genetskim kodom zdrava osoba. Precizne hemijske razlike između patogenih i staničnih izoforma PrP ostaju nepoznate.
Većina slučajeva CJD je sporadične prirode. Zasnovan je na sporadičnoj mutaciji gena PgR. Porodični oblici CJD (10-15% slučajeva) nasljeđuju se autosomno dominantno i rezultat su točkaste mutacije gena PrP. Transmisivi oblik CJD dokumentovan je nakon hirurških intervencija, na primjer, transplantacije rožnice i jatrogene inokulacije priona u pacijenta, kao i upotrebe hormona rasta ekstrahiranih iz hipofize pacijenta s CJD. Ovi slučajevi ukazuju na veoma duge periode period inkubacije CJD. Intenzivno istraživanje i interesovanje medija za ove bolesti poraslo je u U poslednje vreme zbog varijante CJD-a koja se prenosi jedenjem govedine kontaminirane prionima (goveđa spongiformna encefalopatija).
Klinički, CJD karakterizira progresivna demencija, ponekad povezana s kortikalnim smetnje vida i motorni poremećaji - mioklonus, ponekad paraliza mišića i fascikulacije. Smrt obično nastupa u roku od 1-2 godine od pojave prvih simptoma bolesti. EEG se može snimiti karakteristične promene — periodični kompleksi. Dječja verzija CJD manifestira se mentalnim poremećajima, senzornim poremećajima i ataksijom, te progresivnom demencijom. Neuroimaging i druge metode ispitivanja ne otkrivaju specifični znakovi CJD, iako su karakteristične talamičke promjene (izmjerene MRI) opisane kod mnogih pacijenata sa CJD, dijagnoza je potvrđena samo biopsijom ili obdukcijom mozga. Metode liječenja spongiformne encefalopatije nisu razvijene.
Druge demencije uzrokovane degenerativnim lezijama centralnog nervnog sistema razlikuju se od AD po histološkoj slici moždanog tkiva (kod Pickove bolesti). Osim toga, neurodegeneracija kod ovih bolesti, barem na početku bolesti, ograničena je na oštećenje frontalnog i temporalnog režnja (frontotemporalna demencija). Stoga pacijenti doživljavaju demencija frontalni tip sa poremećajima ličnosti društveno ponašanje i više izvršne funkcije, obično povezane s progresivnom dinamičkom disfazijom (koja je rezultat fokalne atrofije frontalnog režnja). Mogući razvoj semantička demencija sa poteškoćama u pronalaženju riječi i gubitkom općeg znanja (kao rezultat fokalne atrofije temporalnog režnja). Tečnost govora sa progresivnom disfazijom je očuvana u ovim slučajevima. Pickova bolest je češća kod mlađih pacijenata.
Demencija može biti povezana s poremećajima kretanja kao što su Huntingtonova bolest i progresivna supranuklearna paraliza. U ovim slučajevima može se uzeti u obzir demencija subkortikalni sa izraženim usporavanjem razmišljanja ( bradifrenija), promjene ličnosti i raspoloženja, te relativno odsustvo kortikalnih deficita (afazija, apraksija i agnozija) koji su tipični za AD.
Neke demencije uzrokovane neurodegeneracijom karakteriziraju se kombinacijom kortikalnih i subkortikalnih manifestacija. posebno, demencija sa Lewyjevim telima(DTL) se smatra jednim od uobičajeni razlozi iscrpljujućih procesa. Lewyjeva tijela - uobičajena histološka karakteristika Parkinsonova bolest povezana je s degeneracijom nigrostriatalnih neurona, ali u DLB su raspoređeni difuznije. Prepoznatljive karakteristike DTL:
- motoričke fluktuacije sa noćnom konfuzijom
- vizuelne halucinacije
- kliničke manifestacije parkinsonizma
- pojačanje simptoma prilikom uzimanja antiparkinsonika i antipsihotika.
Neki pacijenti sa uznapredovalom Parkinsonovom bolešću razviju demenciju nekoliko godina nakon pojave. motoričkih poremećaja(Parkinsonova bolest sa demencijom - PPD). U takvim slučajevima, teško je razlučiti da li pacijent ima AD s pratećom Parkinsonovom bolešću ili difuzno oštećenje neurona mozga s Lewyjevim tijelima. DLB je podržan razvojem poremećaja kretanja i demencije koji su se javili u roku od godinu dana. Za BPD, početak demencije je tipičniji godinu dana ili kasnije nakon razvoja parkinsonizma.
Nedegenerativni uzroci demencije
Multiinfarktna demencija
Uobičajeno stanje uzrokovano rekurentnom tromboembolijom iz ekstrakranijalnih izvora ili, češće, oštećenjem cerebralnih arterija malog kalibra. Pretpostavka o vaskularne prirode potvrđuju slijedeći znakovi:
- akutni početak i postepena progresija, dok kod astme razvoj teče glatko
- vaskularno oštećenje drugih organa i prisustvo vaskularnih faktora rizika
- kombinovani kortikalni i subkortikalni deficit
- noćna konfuzija i subkortikalni poremećaji
- labilnost, fluktuirajuće kognitivno oštećenje
- emocionalna labilnost i manifestacije pseudobulbarne paralize.
Liječenje multiinfarktne demencije ograničeno je na ciljanje vaskularnih faktora rizika kako bi se spriječila progresija bolesti.
Ostali nedegenerativni uzroci demencije navedeni su u tabeli. 4. Mnogi od njih su potencijalno izlječivi. Dva uslova u kojima je to moguće hirurška intervencija, opisani su u nastavku.
Hronični subduralni hematom
Javlja se uglavnom kod starijih ljudi zbog relativno lakša povreda glave. Detalji o povredi mogu nedostajati kako prođu mjeseci ili čak godine prije nego što odete ljekaru. Predispozicija starijih pacijenata za stvaranje subduralnog hematoma posljedica je atrofije mozga i stanjivanja zidova venskih žila u subduralnom prostoru. Manje povrede glave, kao što su one povezane sa zloupotrebom alkohola, mogu uzrokovati obilno krvarenje, posebno kod pacijenata sa poremećajima hemostatskog sistema.
Morfološki, subduralni hematom je šupljina ispunjena žutom ili smeđom tekućinom koja nastaje kao rezultat razaranja oblikovani elementi krv; šupljina je okružena membranom i postepeno se povećava u volumenu. Ranije se vjerovalo da su degradacija proteina i povećanje osmotskog tlaka unutar šupljine glavni mehanizmi za povećanje volumena hematoma, ali trenutno više važan faktor razmatra se ponavljano krvarenje. Klinička slika je u velikoj mjeri određena prisustvom efekta mase sa pomakom moždanih struktura (osim slučajeva obostranih subduralnih hematoma).
Klinički, pacijenti doživljavaju demenciju, kao i fluktuacije u svijesti, epileptičke napade i znakove pojačanog intrakranijalnog pritiska i fokalni neurološki deficiti.
Dijagnoza se obično potvrđuje CT ili MRI glave (slika 4), iako u ranim fazama dijagnostičke poteškoće mogu nastati ako hematom ima istu gustinu kao moždano tkivo, kao i ako postoji obostrani hematom i nema pomaka struktura srednje linije.
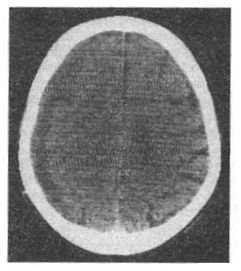
Rice. 4. CT mozga - kronični subduralni hematom
Hidrocefalus normalnog pritiska
Manifestira se klasičnom kliničkom trijadom:
- demencija
- smetnje u hodu
- rana urinarna inkontinencija.
CT snimkom otkriva izraženu dilataciju ventrikula bez kortikalna atrofija. Patogeneza ostaje nejasna. Iako lumbalna punkcija može otkriti normalan krvni tlak, stalno praćenje u roku od 1-2 dana omogućava vam da snimite nekoliko epizoda njegovog povećanja. Rezultati hirurškog ventrikuloperitonealnog ranžiranja mogu varirati, iako se često uočavaju pozitivni rezultati.
Bolesti motornih neurona
Bolest motornih neurona (također poznata kao amiotrofična lateralna skleroza) je progresivna degenerativna lezija kortikalnih, matičnih i spinalnih motornih neurona (na primjer, i CMN i LMN).
Epidemiologija
Stopa incidencije je 2 osobe na 100.000 godišnje. Kod muškaraca postoji predispozicija da ovu bolest(1,5:1), maksimalna prevalencija je među osobama srednje i starije životne dobi, vrhunac pojave bolesti je u šestoj deceniji života. Otprilike 5-10% pacijenata ima porodičnu anamnezu, vjerovatno sa autozomnim načinom nasljeđivanja, a kod takvih pacijenata početak bolesti se opaža kod više od rane godine. U porodičnim slučajevima, mutacija se javlja u genu odgovornom za sintezu enzima superoksid dismutaze.
Etiologija i patogeneza
Dva mehanizma degeneracije motornih neurona smatraju se najvažnijim u patogenezi bolesti:
- ekscitotoksičnost- toksini stupaju u interakciju sa glutamatnim receptorima, što dovodi do viška intracelularnog kalcijuma
- slobodni radikali imaju štetni učinak na motorne neurone, pokrećući niz patogenetskih reakcija.
Ova dva mehanizma mogu djelovati zajedno. Stoga se slobodni kisikovi radikali mogu proizvesti kao odgovor na povećane razine intracelularnog kalcija, čiju akumulaciju, zauzvrat, induciraju neidentificirani ekscitotoksini.
Kliničke manifestacije i prognoza
Paraliza i atrofija mišića gornji udovi razvijaju češće od nižih. Grčevi i fascikulacije mogu prethoditi drugim poremećajima kretanja. Pregledom se otkriva kombinacija znakova oštećenja centralnih i perifernih motornih neurona (CMN i PMN). Dijagnoza je očigledna kada ovi znakovi koegzistiraju u istom području (npr mlitava paraliza i povećani tetivni refleksi u gornjim ekstremitetima), kao i kada je istovremeno zahvaćeno više područja (kranijalni nervi, gornji i donji ekstremiteti) sa znacima progresije bolesti. Dijagnostičke poteškoće nastaju na početku bolesti, kada postoje znaci izoliranog oštećenja centralnog ili perifernog motornog neurona jednog ekstremiteta. Treba napomenuti da 10% pacijenata doživi oštećenje isključivo perifernih motornih neurona u toku bolesti (ranije se ovaj tok nazivao „progresivna mišićna atrofija“).
Poremećaji kretanja obično se šire asimetrično, barem u početku. Nema senzornih poremećaja, kao ni poremećaja sfinktera, osim zatvora uzrokovanog slabošću mišića zdjelice i trbušni zid i smanjen unos tečnosti. Neki pacijenti razvijaju frontalnu demenciju.
Neki pacijenti imaju dizartriju i disfagiju („progresivna bulbarna paraliza"). Postoje znaci mješovite bulbarne i pseudobulbarne paralize - fascikulacija jezika sa živim mandibularnim refleksom. Ovi pacijenti su pod rizikom od plućnih infekcija zbog visokog rizika od aspiracije i slabosti respiratornih mišića. Iste komplikacije se razvijaju kod pacijenata s pretežno zahvaćenim ekstremitetima, od kojih većina kasnije razvije bulbarne simptome. Ostali simptomi bolesti:
- depresija sa sve većom društvenom izolacijom
- gubitak težine, pothranjenost i dehidracija zbog disfagije
- tromboembolija plućna arterija kao rezultat produžene imobilizacije
- poremećaji disanja kao glavni uzrok smrti.
Prosječan životni vijek za bolesti motornih neurona je 4 godine, a prognoza za bolesnike s bulbarnim poremećajima na početku bolesti je nepovoljnija. Samo 10% pacijenata preživi više od 5 godina, većina njih pokazuje znakove izoliranog oštećenja motornih neurona.
Pregled i dijagnoza
Krvni testovi obično ne otkrivaju promjene, osim mogućeg povećanja nivoa kreatin kinaze.
On EMG otkrivaju se znaci denervacije zbog oštećenja ćelija prednjih rogova. Studija nervna provodljivost omogućava nam da isključimo motornu neuropatiju, koja ima sličnu kliničku sliku varijanti bolesti sa znacima isključivog oštećenja perifernog motornog neurona.
Obavezno u dijagnostici teški slučajevi kako bi se izbjegla kompresija kičmene moždine ili kičmenog korijena.
Bulbarni sindrom zahtijeva diferencijalnu dijagnozu s mijastenijom gravis. Za razliku od mijastenije gravis, bolest motornih neurona rijetko utječe na pokrete očiju.
Budući da je prognoza loša, dijagnoza bolesti motornih neurona treba se temeljiti na jasnoj klinički kriterijumi, posebno s obzirom na mogućnost koegzistencije CMN i PMN lezija na više nivoa sa znacima progresije bolesti. S obzirom na mogućnost prisustva potencijalno izlječivih bolesti koje imaju sličnu kliničku sliku, diferencijalna dijagnoza zahtijeva izuzetno pažljiv pristup.
Tretman
Tretman lijekovima
Većina lijekova ima simptomatsko djelovanje:
- Antiholinergici - za smanjenje salivacije kod pacijenata sa poteškoćama pri gutanju (mogući su i drugi pristupi, poput injekcija botulinum toksina u pljuvačne žlijezde).
- Za spastičnost se koriste baklofen, dantrolen, tizanidin i diazepam.
- Kininski preparati za grčeve.
- Antidepresivi.
- Laksativi (zajedno sa povećan unos tečnosti) za zatvor.
- U svrhu simptomatskog ublažavanja dispneje u najtežim slučajevima - opijati, diazepam.
Ekscitotoksična teorija bolesti motornih neurona dovela je do razvoja lijeka riluzol, koji ima antiglutamatni efekat. Postoje dokazi o blagom produženju očekivanog životnog vijeka kod nekih pacijenata s ovom bolešću kao rezultat primjene riluzola.
Druge terapijske mjere
- Fizioterapija.
- Pomoć u komunikaciji kod pacijenata sa dizartrijom.
- Adaptacija na kućne uslove - pomoć radnog terapeuta.
- Savjeti logopeda i nutricionista za disfagiju.
- Može biti potrebna teška disfagija potkožna endoskopska gastrostomija kako bi se ispravili nedostaci gutanja i osigurao adekvatan unos hrane i tekućine.
- Umjetna ventilacija pluća u slučaju respiratornog distresa, na primjer tokom noćnog sna, kada su ostale motoričke funkcije relativno netaknute, međutim, to postavlja etički problem kod pacijenata sa uznapredovalom slikom bolesti – treba li tako bolan život produžiti .
- Možda će biti potrebna bolnička njega.
- Informacije o pacijentima i podrška Udruženja za bolesti motornih neurona.
Nasljedne bolesti nervnog sistema su zbirni naziv za veliku grupu nozoloških bolesti koje nastaju kao posljedica genetski uvjetovanog oštećenja struktura kičmene moždine ili mozga, tkiva perifernih nervnih vlakana i nervnih vlakana odgovornih za inervaciju mišića. .
Postoji mnogo različitih vrsta nasljednih bolesti nervnog sistema, a ove bolesti su češće od drugih urođenih patologija koje se mogu prenijeti s roditelja na djecu. Činjenica je da je ljudski nervni sistem izuzetno ranjiva struktura, pa svaka odstupanja i štetni efekti tokom intrauterinog razvoja, posebno na gene odgovorne za anatomiju i normalno funkcioniranje nervnog sistema, mogu izazvati pojavu patologija.
Koje su nasljedne patologije nervnog sistema?
Simptomi koji prate gotovo sve nasljedne bolesti nervni sistem se, po pravilu, manifestiraju toliko značajno da utječu na cijeli život osobe, čineći ga manje ispunjenim.
Trenutno je poznato više od 500 različitih patologija nervnog sistema, koje mogu dovesti do širokog spektra sindroma i simptomatskih manifestacija. Genetske patologije igraju važnu ulogu u nastanku cirkulacijskih poremećaja mozga i kičmene moždine, migrene, epilepsije, autonomnih paroksizama, mucanja, histerije, distrofije i drugih abnormalnosti koje nastaju kao posljedica oštećenja nervnih vlakana u različitim dijelovima nervnog sustava. sistem.
Postoji mnogo nozoloških oblika patologija, ali svi imaju neke opšti znakovi, a ponekad i etiologije. Sve nasljedne bolesti nervnog sistema su degenerativne i nemaju znakove meningealnog sindroma; posebno, sastav cerebrospinalne tekućine gotovo nikada ne odstupa od norme.
Različite nozološke grupe bolesti nervnog sistema imaju drugačiji tip naslijeđe i može se prenositi na autosomno dominantan, autosomno recesivan i recesivan način. Recesivni put prijenosa patologije znači da je bolest povezana sa spolom. U ovom slučaju, incidencija se opaža samo kod muškaraca, iako žene mogu biti nosioci defektnih gena.
Autosomno dominantni način nasljeđivanja podrazumijeva prijenos bolesti s generacije na generaciju, pa jedan od roditelja ima znakove i simptome bolesti. Autosomno recesivni put prijenosa pretpostavlja da su oba roditelja nosioci defektnih gena, ali nemaju znakove određene bolesti nervnog sistema.
Bolesti nervnog sistema nasljedne prirode odlikuju se značajnim polimorfizmom i mogu imati različite struje, skup simptoma i stepen njihovog intenziteta, čak i kada se posmatraju kod članova iste porodice. Polimorfizam, čak i unutar jedne porodice, posledica je uticaja drugih postojećih gena, spoljašnjih i unutrašnjih faktora na mutirani gen.
U zavisnosti od karakteristične karakteristike sve bolesti se dijele na velike klase. Uprkos činjenici da je trenutno dosta toga sastavljeno puna klasifikacija bolesti nervnog sistema koje se mogu naslijediti, još uvijek se ne zna sve o patogenezi bolesti, što u velikoj mjeri otežava njihovo liječenje. Trenutno je razvijena prilično jasna klasifikacija nasljednih bolesti.
Sistemske degeneracije nervnog sistema nasljedne prirode
 Klasa sistemskih naslednih degenerativnih bolesti nervnog sistema, koje se prenose genetski, obuhvata veliki broj podklase bolesti koje su rezultat oštećenja određenih struktura. Klasa sistemskih degeneracija nervnog sistema uključuje 4 glavne podklase.
Klasa sistemskih naslednih degenerativnih bolesti nervnog sistema, koje se prenose genetski, obuhvata veliki broj podklase bolesti koje su rezultat oštećenja određenih struktura. Klasa sistemskih degeneracija nervnog sistema uključuje 4 glavne podklase.
Bolesti 1. klase uključuju patologe koje karakterizira oštećenje malog mozga i struktura koje pružaju njegove veze. Ove bolesti uključuju spinocerebelarnu ataksiju, uključujući:
- Pierre-Marie ataksija;
- porodična Friedreichova ataksija;
- kasna kortikalna cerebelarna atrofija Marie-Foy-Alajouanin;
- olivocerebelarna atrofija;
- olivo-ponto-cerebelarna degeneracija svih tipova.
Druga podklasa degenerativnih bolesti nervnog sistema uključuje patologije koje karakterizira oštećenje piramidalnih puteva. Takve bolesti uključuju:
- spastična porodična paraplegija Strumpela;
- Ferguson-Critchley multipla skleroza;
- spastična paraplegija praćena degeneracijom retine.
 Sljedeću podklasu predstavljaju bolesti nervnog sistema koje zahvaćaju subkortikalne ganglije. Ove bolesti uključuju veliki broj opasnih patoloških stanja:
Sljedeću podklasu predstavljaju bolesti nervnog sistema koje zahvaćaju subkortikalne ganglije. Ove bolesti uključuju veliki broj opasnih patoloških stanja:
- Parkinsonov sindrom;
- hepatocerebralna distrofija;
- hronična Huntingtonova koreja;
- kalcifikacija bazalnih ganglija;
- drhtanje;
- Gilles de la Touretteov sindrom;
- nasledna progresivna mioklonusna epilepsija;
- dvostruka atetoza;
- nasljedna deformirajuća distonija.
Posljednju podklasu predstavljaju neuroretinalne degeneracije. Upečatljiv primjer bolesti koje pripadaju ovoj podklasi je Leberova nasljedna optička atrofija, kao i Lawrence-Moon-Biedl sindrom.
Nasljedne neuromuskularne bolesti
Nasljedne neuromišićne bolesti predstavljaju najopsežnija grupa patologija.
Ova grupa bolesti poznata je i kao neuromuskularne distrofije (NMD).
 Neuromuskularne distrofije predstavljaju 5 velikih grupa bolesti, od kojih svaka uključuje mnogo različitih nozoloških oblika.
Neuromuskularne distrofije predstavljaju 5 velikih grupa bolesti, od kojih svaka uključuje mnogo različitih nozoloških oblika.
Grupa 1 neuromuskularne distrofije uključuje spinalne amiotrofije. Uobičajeni oblici spinalne amiotrofije uključuju:
- progresivna spinalna Aran-Duchenneova amiotrofija, uočena kod odraslih;
- Kugelberg-Welander juvenilna amiotrofija;
- dečji oblik Werdnig-Hoffmannove amiotrofije.
Drugu grupu neuromuskularne distrofije predstavljaju neuralne amiotrofije. Ove bolesti mogu biti porodične ili sporadične. Ova grupa bolesti najčešće se manifestuje u djetinjstvu ili adolescencija. Ova grupa je nervozna mišićne distrofije vezati:
- Roussy-Lévyjev sindrom;
- neuralni oblik amiotrofije Charcot-Marie-Tooth;
- Refsumova bolest;
- hipertrofična intersticijska neuropatija Dejerine-Sotta.
Veliku grupu neuromišićnih bolesti predstavljaju primarne progresivne mišićne distrofije. Manifestacije primarne progresivne mišićne distrofije mogu biti različite. Istaknuti predstavnici ove grupe bolesti su:
- pseudohipertrofična dječja distrofija Duchenne;
- povoljna trenutna pseudohipertrofična distrofija Becker-Keenera;
- juvenilna i krajnja lumbalna Erbova distrofija;
- distalna mišićna distrofija;
- okulofaringealni i okularni oblici distrofije;
- sindrom krute kralježnice;
- neprogresivna mišićna distrofija.
 Grupa 4 neuromuskularne distrofije uključuje miotoniju. Različiti fenotipovi miotonije nasljeđuju se na autosomno dominantan način i karakteriziraju ih odgođena relaksacija mišića. Uobičajene varijante nasljedne miotonije uključuju:
Grupa 4 neuromuskularne distrofije uključuje miotoniju. Različiti fenotipovi miotonije nasljeđuju se na autosomno dominantan način i karakteriziraju ih odgođena relaksacija mišića. Uobičajene varijante nasljedne miotonije uključuju:
- nasljedna paramiotonija Eulenburg;
- Thomsenova miotonija;
- neuromiotonija;
- distrofična miotonija.
Grupa 5 neuromuskularne distrofije uključuje mioplegične sindrome i paroksizmalnu mioplegiju. Najčešće mioplegije uključuju:
- normokalemijska mioplegija;
- Hamstorp bolest;
- hipokalemijska paroksizmalna mioplegija;
- sekundarne varijante paroksizmalne mioplegije.
Sve ove bolesti imaju svoje karakteristike toka i razvoja. Neki od njih dozvoljavaju bolesnima da žive pun život i ne doživljavaju očiglednu nelagodu, dok drugi dovode do značajnog poremećaja mišićnog sistema.
Nasljedne fakomatoze i bolesti nervnog sistema sa oštećenjem vezivnog tkiva
 Fakomatoze su grupa bolesti povezanih sa ektodermalno-mezodermalnim displazijama. Glavna manifestacija ove grupe bolesti je prisustvo abnormalnosti pigmentacije kože te ozbiljne abnormalnosti nervnog sistema i rada unutrašnje organe. Takve bolesti uključuju:
Fakomatoze su grupa bolesti povezanih sa ektodermalno-mezodermalnim displazijama. Glavna manifestacija ove grupe bolesti je prisustvo abnormalnosti pigmentacije kože te ozbiljne abnormalnosti nervnog sistema i rada unutrašnje organe. Takve bolesti uključuju:
- tuberozna skleroza;
- encefalotrigeminalna angiomatoza;
- Louis-Bar sindrom;
- Hipel-Lindauova sistemska angioretikulomatoza.
Nasljedne bolesti koje pogađaju vezivno tkivo i pogađaju nervni sistem uključuju:
- Marfanov sindrom;
- mukopolisaharidoze;
- sindrom krhkih kostiju.
Sve ove patologije, u jednom ili drugom stepenu, dovode do poremećaja u radu nervnog sistema. U nekim slučajevima uočavaju se manja odstupanja, dok su u drugima poremećaji u radu nervnih vlakana sistemske prirode.
Nasljedne bolesti nervnog sistema praćene metaboličkim poremećajima
 Postoji mnogo nasljednih bolesti koje karakteriziraju ozbiljni poremećaji metabolički procesi u organizmu i dovode do abnormalnosti u funkcionisanju nervnog sistema. Metabolički poremećaji se mogu izraziti patologijama u metabolizmu lipida, aminokiselina, ugljikohidrata, a pored toga, poremećajima ravnoteže bilirubina i pigmenta i cističnom fibrozom. Takve nasljedne bolesti uključuju:
Postoji mnogo nasljednih bolesti koje karakteriziraju ozbiljni poremećaji metabolički procesi u organizmu i dovode do abnormalnosti u funkcionisanju nervnog sistema. Metabolički poremećaji se mogu izraziti patologijama u metabolizmu lipida, aminokiselina, ugljikohidrata, a pored toga, poremećajima ravnoteže bilirubina i pigmenta i cističnom fibrozom. Takve nasljedne bolesti uključuju:
- fenilpiruvična oligofrenija;
- poremećaj metabolizma tirozina;
- alkaptonurija;
- homocistinurija;
- triptofan;
- histidinemija;
- Niemann-Pickova bolest;
- galaktozemija;
- pentozurija;
- fruktozemija;
- kronična nehemolitička žutica;
- ustavna disfunkcija jetre;
- porfirija;
- hiperbilirubinemija;
- amaurotski idiotizam;
- lipodistrofija;
- Tay-Sachsova bolest.
Ove bolesti se mogu prenijeti na različite načine nasleđe. U nekim slučajevima bolest se manifestira u ranoj dobi, dok u drugima manifestacije mogu biti paroksizmalne prirode. Na primjer, porfirija se obično manifestira pod utjecajem nepovoljni faktori, a nakon dugotrajnog napada možda neće biti vidljivog pogoršanja stanja. Kako se porfirija razvija, ona je praćena polaganim odumiranjem moždanih ćelija, zbog čega bolest može dugo vrijeme curenje skriveno.
Nasljedne neuromuskularne bolesti su velika heterogena grupa bolesti zasnovanih na genetski uslovljenim oštećenjima neuromišićnog sistema. Bolesti karakteriziraju slabost mišića, atrofija mišića, poremećaji statičkih i lokomotornih funkcija.
Prilikom postavljanja dijagnoze uzima se u obzir starost pojave prvih simptoma. kliničkih simptoma bolesti, lokalizacija atrofije i priroda širenja miodistrofičnog procesa (uzlazni, silazni, prisutnost ili odsutnost pseudohipertrofije, fascikulacije, poremećaji osjetljivosti, paroksizmi mišićne slabosti), kao i brzina progresije.
Progresivne mišićne distrofije su najveća grupa. U zavisnosti od prirode primarnih promjena, konvencionalno se razlikuju primarni (miopatije) i sekundarni oblici progresivne mišićne distrofije (denervacijske amiotrofije - spinalne i neuralne).
24.1.1. Progresivne mišićne distrofije
Da bi se objasnili uzroci mišićnih distrofija, predloženo je nekoliko hipoteza (neurogena, vaskularna, membranska), razmatrajući mehanizme nastanka progresivnih mišićnih distrofija sa stanovišta primarnog, genetski uslovljenog defekta.
Progresivna Duchenneova mišićna distrofija. Bolest je opisao Duchenne 1853. godine. Učestalost je 3,3 na 100.000 stanovnika, 14 na 100.000 rođenih. Nasljeđuje se recesivno, X-vezano. U velikoj većini slučajeva oboljevaju dječaci. Duchenneova distrofija je povezana s oštećenjem gena odgovornog za proizvodnju distrofina. Prilikom pregleda majki koje su nosioci gena u genetskim konsultacijama (biopsija horionskih resica u 8-9 sedmici), bolest se otkriva kod dječaka. Slučajevi bolesti kod djevojčica su izuzetno rijetki, iako su mogući kod X0 kariotipa, X0/XX, X0/XXX, X0/XXX/XXX mozaicizma i strukturnih abnormalnosti hromozoma.
Patomorfologija. Karakterizira ga degeneracija mišićno tkivo, njegova zamjena masnim i vezivnim tkivom, nekroza pojedinih vlakana.
Kliničke manifestacije. Znakovi bolesti pojavljuju se u prve 1-3 godine života. Već u 1. godini skreće se pažnja na zaostajanje djece u motoričkom razvoju. U pravilu počinju da sjede, ustaju i hodaju sa zakašnjenjem. Pokreti su nezgodni, djeca su u hodu nesigurna, često se spotiču i padaju. U dobi od 2-3 godine javlja se mišićna slabost i patološki zamor mišića, koji se manifestira tijekom fizičke aktivnosti - dugog hodanja, penjanja uz stepenice i promjene u "pačjem" hodu. U ovom periodu skreće se pažnja na osebujnu „stereotipnu“ dinamiku kretanja djece pri ustajanju iz horizontalnog položaja, iz čučećeg položaja ili sa stolice. Ustajanje se odvija u fazama, uz aktivnu upotrebu ruku - "penjanje uz ljestve" ili "penjanje sam". Atrofija mišića je uvijek simetrična. U početku se lokaliziraju u proksimalnim mišićnim grupama donjih ekstremiteta - mišićima karličnog pojasa, bedara, a nakon 1-3 godine brzo se šire u smjeru prema gore do proksimalnih mišićnih grupa gornjih ekstremiteta - ramenog pojasa. , leđni mišići. Kao rezultat atrofije, pojavljuju se lordoza, lopatice u obliku krila i struk "ose". Tipičan, "klasičan" simptom bolesti je pseudohipertrofija mišiće potkoljenice.
Pri palpaciji mišići su gusti i bezbolni. Kod mnogih pacijenata, kao rezultat selektivnog i neujednačenog oštećenja razne grupe mišići, mišićne kontrakture i retrakcije tetiva javljaju se rano. Tonus mišića je smanjen uglavnom u proksimalnim mišićnim grupama. Tetivni refleksi se mijenjaju u različitim sekvencama. U ranoj fazi bolesti nestaju refleksi koljena, kasnije - refleksi mišića bicepsa i tricepsa. Petni (Ahilovi) refleksi ostaju netaknuti dugo vremena.
Smanjena amplituda oscilacija i povećana polifaza.
Jedna od karakterističnih karakteristika Duchenneove mišićne distrofije je kombinacija ovog oblika sa patologijom osteoartikularnog sistema i unutrašnjih organa (kardiovaskularni i neuroendokrini sistem). Osteoartikularne poremećaje karakteriziraju deformiteti kralježnice, stopala i grudne kosti. Radiografija otkriva suženje medularnog kanala i stanjivanje kortikalnog sloja dugih dijafiza tubularnih kostiju.
Kardiovaskularni poremećaji se klinički manifestuju labilnošću pulsa, krvni pritisak, ponekad tupost tonova i proširenje granica srca. EKG snima promjene na miokardu (blok grane snopa i sl.). Neuroendokrini poremećaji se javljaju kod 30-50% pacijenata. Češće od ostalih, primjećuju se Itsenko-Cushingov sindrom i Babinsky-Froelich adiposogenitalna distrofija. Inteligencija mnogih pacijenata je smanjena različitim stepenima.
Protok. Bolest ima brzo progresivni maligni tok. Do 7-10 godina nastaju duboki poremećaji kretanja - izražena promjena u hodu, smanjenje mišićne snage, što značajno ograničava slobodno, neovisno kretanje pacijenata. U dobi od 14-15 godina dolazi do nepokretnosti.
Dijagnostika i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (recesivni X-vezani tip nasljeđivanja), kliničkih egzacerbacija bolesti (rani početak sa 1-3 godine, simetrične atrofije proksimalnih mišićnih grupa koje se razvijaju u uzlaznom smjeru, pseudohipertrofija potkoljenice mišići, teški somatski i neuroendokrini poremećaji, smanjena inteligencija, brz maligni tok bolesti), podaci iz biohemijskih studija (obično rano, od 5. dana života djeteta, povećanje aktivnosti CPK - 30-50 puta više od normalnog) , elektromiografija igle i morfološki rezultati. omogućava identifikaciju primarnog mišićnog (miodistrofičnog) tipa lezije.
Bolest treba razlikovati od Werdnig-Hoffmannove spinalne amiotrofije, rahitisa i kongenitalne dislokacije kuka.
Beckerova progresivna mišićna distrofija. Bolest je opisao Becker 1955. godine. Učestalost nije precizno utvrđena. Nasljeđuje se na X-vezani recesivni način.
Kliničke manifestacije. Prvi znaci bolesti javljaju se u dobi od 10-15 godina, ponekad i ranije. Početni simptomi su slabost mišića, patološki zamor mišića tokom fizičke aktivnosti, pseudohipertrofija mišića lista. Atrofije se razvijaju simetrično. U početku se lokaliziraju u proksimalnim mišićnim grupama donjih ekstremiteta - zdjeličnog pojasa i kukova, a kasnije se šire na proksimalne mišićne grupe gornjih ekstremiteta. Kao rezultat atrofije dolazi do promjena u "pačjem" hodu i kompenzacijskih miopatskih tehnika pri ustajanju. Tonus mišića u proksimalnim mišićnim grupama je umjereno smanjen. Tetivni refleksi ostaju netaknuti dugo vremena; samo se refleksi koljena rano smanjuju. Kardiovaskularni poremećaji su umjereno izraženi. Ponekad se primjećuju kardialgija i blok grane snopa. Endokrini poremećaji se manifestuju ginekomastijom, smanjenim libidom i impotencijom. Inteligencija očuvana.
Protok. Bolest napreduje sporo. Stopa širenja atrofije je niska, a pacijenti ostaju radno sposobni dugo vremena.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (recesivni tip nasljeđivanja vezan za X hromozom), karakteristika kliničkih manifestacija (početak bolesti sa 10-15 godina, atrofija u proksimalnim mišićnim grupama, sporo, preko 10-20 godina). godine, širenje atrofije u uzlaznom pravcu, masivna pseudohipertrofija mišića lista, umjerena somatskih poremećaja, spor tok), podaci iz biohemijskih studija (povećana aktivnost CPK, LDH u krvi), elektromiografija iglom i morfološki rezultati, koji omogućavaju identifikaciju primarnog mišićnog tipa promjena.
Bolest treba razlikovati od progresivne mišićne distrofije Duchenne, Erb-Roth i spinalne amiotrofije Kugelberg-Welander.
Progresivna Dreyfusova mišićna distrofija. Bolest je opisao Dreyfus 1961. godine. Učestalost nije utvrđena. Nasljeđuje se na X-vezani recesivni način.
Kliničke manifestacije. Prvi znaci bolesti javljaju se u dobi od 5-7 godina. Kao i kod drugih oblika progresivne mišićne distrofije, početak bolesti karakterizira slabost mišića i patološki zamor mišića tijekom fizičke aktivnosti. Atrofije se javljaju simetrično i u početku su lokalizirane u proksimalnim mišićnim grupama donjih ekstremiteta - karličnog pojasa, bedara. Proksimalne mišićne grupe gornjih udova su uključene u miodistrofični proces mnogo kasnije. Posebne karakteristike ovog oblika su rane kontrakture u zglobovima laktova i retrakcija Ahilove tetive. Mnogi pacijenti imaju srčane aritmije. Inteligencija očuvana.
Protok. Bolest napreduje sporo.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (recesivni X-vezani tip nasljeđivanja), kliničkih karakteristika (početak bolesti u dobi od 5-7 godina, simetrične atrofije sa inicijalnom lokalizacijom u proksimalnim mišićnim grupama donjeg i naknadno sa polaganim širenjem miodistrofija na proksimalne mišićne grupe gornjih ekstremiteta, ranim kontrakturama zglobova laktova, povlačenjem Ahilove tetive, kardiovaskularnim poremećajima u vidu aritmija srčane aktivnosti, sporog, progresivnog toka), podaci iz biohemijskih studije (visoka aktivnost CPK), elektromiografija i morfološki podaci koji omogućavaju identifikaciju primarnih promjena mišićnog karaktera.
Bolest treba razlikovati od progresivne mišićne distrofije Becker, Duchenne, Erb-Roth i spinalne amiotrofije Kugelberg-Welander.
Erb-Roth progresivna mišićna distrofija. Učestalost 1,5 na 100.000 stanovnika. Nasljeđuje se autosomno recesivno.
Patomorfologija. Odgovara primarnoj leziji mišića.
Kliničke manifestacije. Prvi znaci bolesti javljaju se uglavnom u dobi od 14-16 godina, izuzetno rijetko u dobi od 5-10 godina. Početni simptomi su slabost mišića, patološki zamor mišića tokom fizičke aktivnosti i promjene u pačjem hodu. Atrofija na početku bolesti lokalizirana je u proksimalnim mišićnim grupama donjih ekstremiteta. Ponekad miodistrofični proces istovremeno zahvaća mišiće zdjelice i ramenog pojasa. U mnogo kasnijim fazama u proces su uključeni mišići leđa i abdomena. Kao rezultat atrofije, pojavljuju se lordoza, lopatice u obliku krila i struk "ose". Prilikom ustajanja, pacijenti koriste pomoćne tehnike - ustajanje sa „merdevinama“. Pseudohipertrofija mišića, kontrakture zglobova i retrakcije tetiva obično su umjereno izraženi. Već u ranoj fazi bolesti tipično je smanjenje refleksa koljena i refleksa bicepsa i tricepsa brachii mišića.
Protok. Bolest brzo napreduje. Invaliditet se javlja rano.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu podataka genealoške analize (autosomno recesivni tip nasljeđivanja), kliničkih karakteristika (početak bolesti uglavnom u dobi od 14-16 godina, atrofija proksimalnih mišićnih grupa, umjerena pseudohipertrofija, brza progresija), rezultata elektromiografija igle i morfološki podaci koji omogućavaju identifikaciju primarne mišićne prirode promjena.
Bolest treba razlikovati od progresivne Beckerove mišićne distrofije i Kugelberg-Welanderove spinalne amiotrofije.
Landouzy-Dejerine skapulohumeralno-facijalni oblik. Bolest su opisali Landouzi i Dejerine 1884. godine. Učestalost je 0,9-2 na 100.000 stanovnika. Nasljeđuje se autosomno dominantno.
Kliničke manifestacije. Prvi znaci se javljaju uglavnom u dobi od 10-20 godina. Slabost i atrofija mišića lokalizirani su u području mišića lica, lopatica i ramena. Zbog atrofije lice postaje hipomimično. Tipični su „uglačano“ čelo, lagoftalmus, „poprečni“ osmeh, debele, ponekad izbočene usne („tapir usne“). Atrofija mišića bicepsa i tricepsa ramena, pectoralis major, serratus anterior i trapezius mišića uzrokuje pojavu simptoma labavih ramenih pojaseva, lopatica u obliku krila, pojavu širokog međulopatičnog prostora, spljoštenost prsa, skolioza. U nekim slučajevima, atrofija se širi na mišiće nogu (facioscapulohumeral, facioscapulohumeral-humeroperoneal, facioscapulohumeral-humeral-femoral, facioscapulohumeral-femoral-peroneal i druge opcije). Pseudohipertrofija je izražena u mišićima lista i deltoidnih mišića. Tonus mišića u ranoj fazi bolesti je smanjen u proksimalnim mišićnim grupama. Refleksi tetiva su smanjeni uglavnom u mišićima bicepsa i tricepsa brachii.
Protok. Tipično, bolest napreduje sporo. Pacijenti ostaju funkcionalni dugo vremena.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (autosomno dominantni tip nasljeđivanja), kliničkih karakteristika (uglavnom glenohumeralno-facijalna lokalizacija miodistrofičnog procesa).
Bolest treba razlikovati od drugih progresivnih mišićnih distrofija: Erb-Roth, Becker.
24.1.2. Neurogene amiotrofije
Spinalna amiotrofija Werdnig-Hoffmanna. Bolest su opisali J. Werdnig 1891. i J. Hoffmann 1893. Učestalost je 1 na 100.000 stanovnika, 7 na 100.000 novorođenčadi. Nasljeđuje se autosomno recesivno.
Patomorfologija. Otkrivaju se nerazvijenost ćelija prednjih rogova kičmene moždine i demijelinizacija prednjih korijena. Često postoje slične promjene u motoričkim jezgrima i korijenima V, VI, VII, IX, X, XI i XII kranijalni nervi. U skeletnim mišićima, neurogene promjene karakterizira "atrofija fascikula", izmjena atrofiranih i intaktnih fascikula mišićna vlakna, kao i poremećaji tipični za primarne miopatije (hialinoza, hipertrofija pojedinačnih mišićnih vlakana, hiperplazija vezivnog tkiva).
Kliničke manifestacije. Postoje tri oblika bolesti: kongenitalni, rani dječji i kasni, koji se razlikuju po vremenu ispoljavanja prvih kliničkih simptoma i tempu amiotrofičnog procesa.
Kod kongenitalnog oblika, u prvim danima života, djeca doživljavaju generaliziranu mišićnu hipotoniju i gubitak mišića, smanjene ili izostanke tetivnih refleksa. Bulbarni poremećaji se otkrivaju rano, manifestiraju se usporenim sisanjem, slabim plačem, fibrilacijama jezika i smanjenim faringealnim refleksom. Bolest se kombinuje sa osteoartikularnim deformitetima: skolioza, levkasti grudni koš ili "pileći" grudni koš, kontrakture zglobova. Razvoj statičkih i lokomotornih funkcija naglo je usporen. Samo ograničen broj djece razvija sposobnost samostalnog držanja glave i sjedenja sa značajnim zakašnjenjem. Međutim, stečene motoričke sposobnosti brzo nazaduju. Mnoga djeca s urođenim oblikom bolesti imaju smanjenu inteligenciju. Često posmatrano urođene mane razvoj: kongenitalni hidrocefalus, kriptorhizam, hemangiom, displazija zglobovi kuka, klupsko stopalo itd.
Protok. Bolest ima brzo progresivni tok. Smrt nastupa prije navršenih 9 godina. Jedan od glavnih uzroka smrti su teški somatski poremećaji (kardiovaskularni i respiratorna insuficijencija), uzrokovano slabošću mišića prsnog koša i smanjenjem njegovog učešća u fiziologiji disanja.
U ranom dječjem obliku, prvi znaci bolesti pojavljuju se po pravilu u drugoj polovini života. Motorički razvoj u prvim mjesecima je zadovoljavajući. Djeca počinju blagovremeno da drže podignute glave, sjede, a ponekad i stoje. Bolest se razvija subakutno, često nakon infekcije ili trovanja hranom. Flakcidna pareza se u početku lokalizira na nogama, a zatim se brzo širi na mišiće trupa i ruku. Difuzna mišićna atrofija je u kombinaciji sa fascikulacijama, fibrilacijama jezika, finim tremorom prstiju i kontrakturama tetiva. Mišićni tonus, tetivni i periostalni refleksi su smanjeni. U kasnijim fazama javlja se generalizirana mišićna hipotonija i simptomi bulbarne paralize.
Protok. Maligni, iako blaži u odnosu na kongenitalni oblik. Smrt nastupa u dobi od 14-15 godina.
U kasnoj formi, prvi znaci bolesti pojavljuju se u dobi od 1,5-2,5 godine. Do ove dobi djeca su u potpunosti završila formiranje statičkih i lokomotornih funkcija. Većina djece samostalno hoda i trči. Bolest počinje neopaženo. Pokreti postaju nespretni i nesigurni. Djeca se često spotaknu i padaju. Hod se mijenja: hodaju sa savijenim nogama u koljenima (hod “lutke na navijanje”). Flakcidna pareza je u početku lokalizirana u proksimalnim mišićnim grupama donjih ekstremiteta, zatim se relativno polako pomiče na proksimalne mišićne grupe gornjih ekstremiteta i mišiće trupa; mišićna atrofija je obično suptilna zbog dobro razvijenog potkožnog masnog sloja. Tipični su fascikulacije, fini tremor prstiju, bulbarni simptomi - fibrilacija i atrofija jezika, smanjeni faringealni i palatinalni refleksi. Tetivni i periostalni refleksi nestaju već u ranim stadijumima bolesti. Osteoartikularni deformiteti se razvijaju paralelno sa osnovnom bolešću. Najizraženija deformacija grudnog koša.
Protok. Maligni, ali blaži od prva dva oblika. Oštećenje sposobnosti samostalnog hodanja javlja se u dobi od 10-12 godina. Bolesnici žive do 20-30 godina.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se zasniva na genealoškoj analizi (autosomno recesivni tip nasljeđivanja), kliničkim karakteristikama (rani početak, prisutnost difuzne atrofije s pretežnom lokalizacijom u proksimalnim mišićnim grupama, generalizirana mišićna hipotonija, fascikulacije i fibrilacije jezika, odsustvo pseudohipertrofija, a u većini slučajeva maligni tok i sl.), rezultati globalne (kožne) i igličaste elektromiografije i morfološkog pregleda skeletnih mišića, što nam omogućava da identifikujemo denervacioni karakter promjena.
Razlikovati urođene i rani oblik proizilazi prvenstveno iz bolesti koje spadaju u grupu sindroma sa kongenitalnom mišićnom hipotonijom (sindrom „flacidnog djeteta”): Oppenheimova amiatonija, kongenitalni benigni oblik mišićne distrofije, atonički oblik cerebralne paralize, nasljedne metaboličke bolesti, hromozomski sindromi bi trebali itd. razlikovati od spinalne amiotrofije Kugelberg-Welander, progresivne mišićne distrofije Duchennea, Erb-Rotha, itd.
Tretman. Za amiotrofiju kičme Werdnig-Hoffmanna propisana je terapija vježbanjem, masaža i lijekovi koji poboljšavaju trofizam nervnog tkiva– Cerebrolizin, aminalon (gamalon), piriditol (encefabol).
Spinalna juvenilna pseudomiopatska mišićna atrofija Kugelberg-Welander.
Patomorfologija. Otkrivaju se nerazvijenost i degeneracija ćelija u prednjim rogovima kičmene moždine, demijelinizacija prednjih korijena i degeneracija motoričkih jezgara IX, X i XII kranijalnih živaca. U skeletnim mišićima postoje kombinovane promene tipične za neurogenu amiotrofiju (fascikularna atrofija mišićnih vlakana) i primarne miodistrofije (atrofija i hipertrofija mišićnih vlakana, hiperplazija vezivnog tkiva).
Kliničke manifestacije. Prvi znaci bolesti javljaju se u dobi od 4-8 godina. Opisani su slučajevi pojave bolesti u kasnijoj dobi – 15-30 godina. Na početku bolesti karakteristični simptomi su patološki zamor mišića u nogama tokom duže fizičke aktivnosti (hodanje, trčanje), a ponekad i spontani trzaji mišića.
Izvana, povećani mišići lista privlače pažnju. Atrofije su u početku lokalizirane u proksimalnim mišićnim grupama donjih ekstremiteta, karličnog pojasa, bedara i uvijek su simetrične. Njihova pojava uzrokuje ograničenje motoričkih funkcija u nogama - poteškoće pri penjanju uz stepenice, ustajanju s horizontalne površine. Hod se postepeno mijenja. U fazi izraženih poremećaja kretanja poprima „patkiji“ karakter. Atrofije u proksimalnim mišićnim grupama gornjih ekstremiteta obično se razvijaju nekoliko godina nakon oštećenja donjih ekstremiteta. Zbog atrofije lopatične i ramene regije, opseg aktivnih pokreta u rukama se smanjuje, lopatice postaju „krilaste“. Tonus mišića u proksimalnim mišićnim grupama se smanjuje. Refleksi tetiva blede prvo u nogama, a zatim u rukama (refleksi mišića bicepsa i tricepsa brachii). Karakteristični simptomi koji razlikuju spinalnu amiotrofiju Kugelberg-Welandera od fenotipski slične primarne progresivne mišićne distrofije Erb-Rotha su mišićna fiskulacija, fibrilacija jezika i fini tremor prstiju. Poremećaji kostiju i zglobova, retrakcije tetiva su umjereni ili ih nema.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (autosomno recesivno, autosomno dominantno, recesivno X-vezano nasljeđe), kliničkih karakteristika (početak bolesti uglavnom u dobi od 4-8 godina, simetrična atrofija mišića, širenje duž uzlazni tip mišićne fascikulacije, mali tremor jezika, pseudohipertrofija mišića potkoljenice, spori progresivni tok), rezultati globalne i igličaste elektromiografije i morfološke studije skeletnih mišića, što nam omogućava da identificiramo denervaciju prirode promjena.
Bolest treba razlikovati od progresivne mišićne distrofije Becker, Erb-Roth i spinalne amiotrofije Werdnig-Hoffmanna.
Nasljedna distalna spinalna amiotrofija. Frekvencija nije podešena. Nasljeđuje se autosomno recesivno, rjeđe - autosomno dominantnim, X-vezanim recesivnim tipom.
Patomorfologija. Odgovara drugoj spinalnoj amiotrofiji m.
Kliničke manifestacije. Prvi znaci bolesti javljaju se uglavnom u prvoj deceniji života. Početni simptomi bolesti su slabost i atrofija distalnih mišića nogu. U 25% slučajeva uočava se slabost i atrofija distalnih mišića ruke. Prepoznatljive karakteristike– grube deformacije stopala, rani gubitak Ahilovog refleksa uz očuvanje koljena i dubokih refleksa šaka, odsustvo senzornih poremećaja.
Protok. Bolest napreduje sporo.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (autosomno dominantni, autosomno recesivni, X-vezani recesivni tip nasljeđivanja), karakteristika kliničke slike (početak u prvoj deceniji života, dominantna lokalizacija atrofije u distalnim dijelovima donjih ekstremiteta, grubih deformiteta stopala, odsustva senzornih poremećaja, sporog napredovanja miodistrofičnog procesa), rezultati globalne i igličaste elektromiografije, što omogućava identifikaciju zahvaćenosti prednjih rogova kičmene moždine u procesu.
Bolest treba razlikovati od distalne Govers-Welanderove miopatije i Charcot-Marie-Tooth neuralne amiotrofije.
Neuralna amiotrofija Charcot-Marie-Tootha. Učestalost 1 na 50.000 stanovnika. Nasljeđuje se na autosomno dominantan način, rjeđe - na autosomno recesivni X-vezani tip.
Patomorfologija. U nervima se otkriva segmentna demijelinizacija, a u mišićima - denervacija sa fenomenima "snopne" atrofije mišićnih vlakana.
Kliničke manifestacije. Prvi znaci bolesti često se javljaju u dobi od 15-30 godina, rjeđe u predškolskoj dobi. Na početku bolesti karakteristični simptomi su slabost mišića i patološki umor u distalnim dijelovima donjih ekstremiteta. Pacijenti se brzo umaraju kada dugo stoje na jednom mjestu i često pribjegavaju hodanju u mjestu (“simptom tramping”) kako bi smanjili umor mišića. Rjeđe, bolest počinje senzornim poremećajima - bol, parestezija, osjećaj puzanja. Atrofije se u početku razvijaju u mišićima nogu i stopala. Atrofija mišića je obično simetrična. Zahvaćeni su peronealna mišićna grupa i prednji mišić tibialis. Usljed atrofije, noge se u distalnim dijelovima oštro sužavaju i poprimaju oblik “obrnute boce” ili “noge rode”. Stopala se deformišu, „izgrizu“, sa visokim lukom. Pareza stola mijenja hod pacijenata. Hodaju sa visoko podignutim nogama: hodanje na petama je nemoguće. Atrofije u distalnim dijelovima ruku – mišićima thenar i hipotenara, kao i u malim mišićima šaka, javljaju se nekoliko godina nakon razvoja amiotrofičnih promjena na nogama. Atrofije na rukama su simetrične. U teškim slučajevima, sa teškom atrofijom, ruke poprimaju oblik "kandžastih", "majmunskih". Tonus mišića je jednoliko smanjen u distalnim udovima. Refleksi tetiva se mijenjaju neravnomjerno: Ahilovi refleksi se smanjuju u ranim fazama bolesti, ali refleks koljena, refleksi mišića tricepsa i bicepsa brachii ostaju dugo netaknuti. Senzorne poremećaje definišu poremećaji površinske osjetljivosti perifernog tipa („tip rukavica i čarapa“). Često postoje vegetativno-trofični poremećaji - hiperhidroza i hiperemija šaka i stopala. Inteligencija je obično očuvana.
Protok. Bolest napreduje sporo. Prognoza je u većini slučajeva povoljna.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se zasniva na genealoškoj analizi (autosomno dominantni, autosomno recesivni, X-vezani recesivni tip nasljeđivanja), kliničkim karakteristikama (atrofija distalnih udova, poremećaji osjetljivosti polineuritičkog tipa, sporo progresivni tok), rezultatima globalne, iglene i stimulacijske elektromiografije (smanjenje brzine provođenja duž senzornih i motornih vlakana perifernih nerava) i, u nekim slučajevima, biopsija živca.
Bolest treba razlikovati od distalne Govers-Welanderove miopatije, nasljedne distalne spinalne amiotrofije, miotonične distrofije, perifernih neuropatija, intoksikacije, infektivnog polineuritisa i drugih bolesti.
Tretman. Terapija progresivnih neuromišićnih bolesti usmjerena je na poboljšanje trofizma mišića, kao i na provođenje impulsa duž nervnih vlakana.
U cilju poboljšanja trofizma miševa propisuju se adenozin trifosforna kiselina, kokarboksilaza, cerebrolizin, riboksin, fosfaden, karnitin hlorid, metnonin, leucin, glutaminska kiselina. Anabolički hormoni se propisuju samo u kratkim kursevima. Koriste se vitamini E, A, grupe B i C. Indikovani su proizvodi koji poboljšavaju mikrocirkulaciju: nikotinska kiselina, ksantinol nikotinat, nikošpan, pentoksifilin, parmidin. Za poboljšanje provodljivosti propisuju se antiholinesterazni lijekovi: galantamin, oksazil, piridostigmin bromid, stefaglabrin sulfat, amiridin.
Uz terapiju lijekovima koristi se fizikalna terapija. masaža i fizioterapija. Važna je prevencija osteoartikularnih deformiteta i kontraktura udova.
IN kompleksan tretman pacijenti koriste sljedeće vrste fizioterapije: elektroforeza lijekovi(prozerin, kalcijum hlorid), dijadinamičke struje, miostimulacija sinusoidnim modulisanim strujama, električna stimulacija nerava, ultrazvuk, ozokerit, blatne aplikacije, radonske, četinarske, sulfidne i sumporovodične kupke, baroterapija kiseonikom. Ortopedsko liječenje je indicirano kod kontraktura udova, umjerenog deformiteta kičme i asimetričnog skraćivanja udova. Indikovani su kompletni proteini, dijeta sa kalijumom, vitamini.
Liječenje treba biti individualno, sveobuhvatno i dugotrajno, koje se sastoji od uzastopnih kurseva, uključujući kombinaciju različitih vrsta terapije.
24.1.3. Paroksizmalna mioplegija
Nasljedna paroksizmalna mioplegija je grupa neuromišićnih bolesti koje karakteriziraju iznenadni napadi mišićne slabosti i plegije. Najčešći nasljedne paroksizmalne mioplegije su hipo-, hiper- i normokalemični oblici. Patogeneza je nejasna. Pretpostavlja se genetski determinisan defekt sarkolemalne membrane, koji narušava permeabilnost za jone natrijuma i kalija,
Hipokalemijski oblik paroksizmalne mioplegije (Westphalova bolest). Bolest je opisao Westphal 1895. Nasljeđuje se autosomno dominantno.
Kliničke manifestacije. Bolest se manifestuje u dobi od 6-15 godina. Paroksizme karakteriziraju iznenadni, noćni ili jutarnjim satima razvoj mišićne slabosti, nepokretnosti, smanjenog mišićnog tonusa, tetivnih refleksa, autonomni poremećaji– labilnost pulsa, krvni pritisak, hiperhidroza. Napadi mogu biti parcijalni, koji uključuju malu grupu mišića, ili generalizirani. U toku napada nastaju poremećaji u kardiovaskularnoj aktivnosti: sistolni šum, promjene EKG-a. Svest je uvek očuvana. Prosječno trajanje napada je nekoliko sati; izuzetno rijetko paroksizmi traju nekoliko dana. Sadržaj kalijuma u krvi tokom napada je manji od 2 mmol/l ili niži. Učestalost napada je promjenjiva. Provociraju ih prejedanje hranom bogatom ugljikohidratima, rashlađivanje i fizička aktivnost.
Tretman. Ishrana bogata kalijumom (šljive, suve kajsije, krompir, suvo grožđe). Za ublažavanje napada oralno se propisuje 10% otopina kalijevog hlorida (1 supena kašika svakih sat vremena) ili 0,5% rastvor u izotoničnom rastvoru natrijum hlorida intravenozno (2-2,5 g na 500 ml rastvora tokom jednog sata). Također je preporučljivo primijeniti panangin intravenski.
Hiperkalemijski oblik paroksizmalnog mioplegina (Gamstorpova bolest). Bolest je opisao I.Gamstorp 1956. godine. Nasljeđuje se autsomno dominantno.
Kliničke manifestacije. Bolest se manifestuje u dobi od 1-5 godina. Simptomi su slični paroksizmima u hipokalemijskom obliku i karakteriziraju ih nagli razvoj mišićne slabosti, plegije, sniženog mišićnog tonusa, tetivnih refleksa i autonomnih poremećaja. Za razliku od hipokalemijske, hiperkalemijska paraliza se obično razvija tokom dana, praćena je jakom parestezijom, kombinovana je sa slabošću mišića lica i artikulacionog aparata i kraće traje (30-40 minuta). Tokom napada, sadržaj kalija u krvi raste na 6-7 mmol/l. Učestalost napada je promjenjiva: od dnevnog do nekoliko puta mjesečno. U interiktalnom periodu nema neuroloških simptoma. Provocirajući faktori su post, fizičke vežbe, što uzrokuje umor.
Tretman. Dijeta sa povećan sadržaj ugljeni hidrati, kuhinjska so, ograničena količina kalijum 40 ml 40% rastvora glukoze se daje intravenozno zajedno sa insulinom subkutano; 20 ml 10% rastvora kalcijum hlorida intravenozno.
Normokalemijska (periodična) paraliza. Nasljeđuje se autosomno dominantno.
Kliničke manifestacije. Bolest se manifestuje prije 10. godine života. Njegova posebnost je relativno sporo (u toku nekoliko dana) paroksizmalno rastuća umjerena slabost mišića trupa, udova i žvačnih mišića, kao i polagana (1-2 tjedna) regresija simptoma. Provocirajući faktori su produženi san, produženi boravak u jednom položaju, hipotermija.
Tretman. Diet rich kuhinjska so. Propisan je acetazolamid (Diacarb).
Protok. Svi oblici paroksizmalne mioplegije sporo napreduju. Prognoza uz pravovremenu dijagnozu, hitne mjere i diferencirana terapija lijekovima povoljno.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize, karakteristika kliničke slike, uzimajući u obzir starost u kojoj bolest počinje, vrijeme nastanka paroksizma (noću, ujutro, danju, u neodređeno vrijeme), ozbiljnost mišićne slabosti, učestalost i trajanje napada, provocirajući faktori, laboratorijski podaci biohemijskog istraživanja (sadržaj bioelektrične aktivnosti mišića).
Bolest treba razlikovati od mioplegije koja nastaje kao posljedica primarnih endokrinih bolesti - tireotoksikoze, Connove bolesti (primarni hiperaldosteronizam), Addisonove bolesti itd.
Tretman. Indicirana je dijeta bogata kuhinjskom soli. Diakarb je propisan.
24.1.4. Miotonija
Miotonija je heterogena grupa neuromuskularnih bolesti, ujedinjenih zajedničkim karakterističnim kompleksom poremećaja mišićnog tonusa, koji se manifestuje teškoćama u opuštanju mišića nakon aktivne kontrakcije.
Postoje nasljedne miotonije (stacionarne, sporo progresivne i periodične, rekurentne forme) i miotonični sindromi.
Kongenitalna miotonija (Leiden-Thomsenova bolest). Bolest je prvi opisao Leiden 1874. Thomsen je 1876. godine skrenuo pažnju na nasljednu prirodu bolesti na primjeru svoje porodice (djeca i brojna rodbina - 20 članova njegove porodice u 4 generacije patilo je od miotonije).
Učestalost 0,3-0,7 na 100.000 stanovnika. Nasljeđuje se autosomno dominantno. Penetracija je veća kod muškaraca.
Patogeneza. Važni su poremećaji u permeabilnosti ćelijske membrane, promene jonske i posredničke razmene (poremećaji u funkcionalnom odnosu u vezi kalcijum-troponin-aktomiozin), povećana osetljivost tkiva na acetilholin i kalijum.
Patomorfologija. Svetlosna mikroskopija otkriva hipertrofiju pojedinačnih mišićnih vlakana; histokemijski se utvrđuje smanjenje veličine mišićnih vlakana tipa II; Elektronska mikroskopija otkriva umjerenu hipertrofiju sarkoplazmatskog retikuluma, promjene u obliku i povećanje veličine mitohondrija, te proširenje telofragme miofibrilarnih vlakana.
Kliničke manifestacije. Prvi put se simptomi bolesti javljaju uglavnom u dobi od 8-15 godina. Vodeći znakovi su miotonični grčevi - poteškoće u opuštanju mišića nakon aktivne napetosti. Miotonični grčevi su lokalizovani u različitim mišićnim grupama, najčešće u mišićima šake, nogu, žvačnim mišićima i kružni mišići oči. Snažno stiskanje prstiju, produžena statička napetost nogu, zatvaranje čeljusti i zatvaranje očiju izazivaju tonične grčeve. Faza opuštanja mišića je odgođena za dugo vrijeme, a pacijenti nisu u stanju brzo otkinuti ruke, promijeniti položaj nogu, otvoriti usta, oči. Ponavljani pokreti smanjuju miotonične grčeve. Povećanje mehaničke ekscitabilnosti mišića utvrđuje se posebnim tehnikama: kada neurološki čekić udari u eminenciju prvog prsta, prinosi se na ruku (od nekoliko sekundi do minute) - "simptom palca"; kada udarni čekić udari u jezik, na njemu se pojavi rupa, stezanje - "jezični simptom" Izgled pacijenata je neobičan. Zbog difuzne hipertrofije različitih mišića podsjećaju na profesionalne sportaše. Pri palpaciji mišići su gusti i tvrdi, ali je objektivno smanjena mišićna snaga. Tetivni refleksi su normalni, ali u teškim slučajevima su smanjeni.
Protok. Bolest napreduje sporo. Radni kapacitet se održava dugo vremena.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se zasniva na genealoškoj analizi (autosomno dominantni tip nasljeđivanja), kliničkim karakteristikama (atletski tip tijela, difuzna mišićna hipertrofija, miotonični sindrom) i podacima globalne elektromiografije (miotonična reakcija).
Bolest treba razlikovati od drugih oblika miotonije, ponekad od pseudohipertrofičnih oblika progresivne mišićne distrofije.
Tretman. Prepisati difenin (0,1-0,2 g 3 puta dnevno tokom 2-3 nedelje), diakarb (0,125 g 2 puta dnevno tokom 2-3 nedelje), preparate kalcijuma (intravenski 10% rastvor kalcijum hlorida 10 ml ili kalcijum glukonat intramuskularno). Pretpostavlja se da difenin ima inhibitorni efekat na mono- i polisinaptičku provodljivost u centralnom nervnom sistemu, a dijakarb menja permeabilnost membrane. Preporučuju se fizioterapija u vidu galvanske kragne i gaćica sa kalcijumom, te terapeutske vježbe.
Distrofična miotonija Rossolimo-Steinert-Kurshman. Bolest je prvi opisao G.I. Rossolimo 1901. godine, a potom Steinert i Kurshman 1912. godine. Učestalost je 2,5-5 na 100.000 stanovnika. Nasljeđuje se autosomno dominantno.
Patogeneza. Nejasno. Sumnja se na primarni defekt membrane.
Patomorfologija. Pomoću svjetlosne mikroskopije otkriva se kombinacija atrofiranih i hipertrofiranih mišićnih vlakana, proliferacija vezivnog tkiva i zamjena mišićnog tkiva masnim i vezivnim tkivom. Elektronska mikroskopija utvrđuje promjene u veličini mitohondrija, destrukciju miofibrilarnog aparata i sarkoplazmatskog retikuluma.
Kliničke manifestacije. Prvi znaci bolesti javljaju se u dobi od 10-20 godina. Karakterizira ga kombinacija miotoničnih, miopatskih, neuroendokrinih, kardiovaskularnih poremećaja. Kompleks miotoničnih simptoma, kao i kod Thomsenove kongenitalne miotonije, manifestuje se miotonskim grčevima i povećanom mehaničkom ekscitabilnosti. Ozbiljnost miotonične pojave u kasnijim fazama bolesti s teškom mišićnom distrofijom slabi. Miopatski sindrom karakterizira patološki zamor mišića, slabost, mišićna atrofija, koji su uglavnom lokalizirani u mišićima lica, vrata i distalnih udova. Zbog atrofije izgled Izgled bolesnika je osebujan: glava je spuštena do vrata, lice je prijateljsko, mršavo, posebno u temporalnim predjelima, kapci su poluspušteni, noge i ruke sužene u distalnim dijelovima. Tipične su “pojedene” noge i “majmunske” ruke. Hod je peronealni (“steppage”), ponekad sa atrofijom proksimalnih mišićnih grupa sa komponentom “patka”. Tonus mišića je smanjen, refleksi tetiva rano blijede. Neuroendokrini poremećaji su raznoliki. Najizraženije promjene su na spolnim žlijezdama. Kod muškaraca se često opaža kriptorhizam, smanjen libido i impotencija; kod žena poremećaji menstrualnog ciklusa. Mnogi pacijenti iskuse rano ćelavost, stanjivanje i suhu kožu. Kardiovaskularni poremećaji su konstantni. Postoji potpuna ili djelomična blokada grana snopa, nizak napon na EKG-u i aritmija.
Bolest napreduje sporo.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza se postavlja na osnovu genealoške analize (autosomno dominantni tip nasljeđivanja), kliničkih karakteristika (kombinacija miotoničnih, miopatskih, neuroendokrinih, kardiovaskularnih poremećaja), rezultata globalne elektromiografije (miotonična reakcija), biohemijskih testova krvi (inzulinska rezistencija).
Bolest treba razlikovati od Thomsenove kongenitalne miotonije, drugih miotoničnih oblika, progresivnih mišićnih distrofija - distalne miopatije, neuralne amiotrofije.
Tretman. Kao i kod kongenitalne miotonije, difenin i dijakarb imaju pozitivan učinak. Indikovana je upotreba anaboličkih steroida (retabolil, nerobol, metilandrostenediol). Sadržaj kalija u ishrani treba smanjiti.
24.2. Piramidalne i ekstrapiramidalne degeneracije
24.2.1. Strumpellova porodična spastična paraliza
Hronična progresivna nasledna degenerativna bolest nervnog sistema, koju karakteriše bilateralno oštećenje piramidalnih puteva u bočnim i prednjim moždinama kičmene moždine. A. Shtrumpel je 1866. godine zabilježio porodičnu prirodu bolesti. Koristi se i naziv "porodična spastična paraplegija Erb-Charcot-Strumpela".
Etiologija i patogeneza. Bolest je nasljedna, najčešće se prenosi autosomno dominantnim, rjeđe autosomno recesivnim i spolno vezanim (X-hromozom) tipom. Patogeneza degeneracije i primarni biohemijski defekt su nepoznati.
Patomorfologija. Najčešće su zahvaćeni lumbalni i torakalni dijelovi kičmene moždine, a rjeđe moždano stablo. Postoji simetrična glijalna degeneracija piramidalnih puteva u bočnim i prednjim vrpcama i Gaulleovim snopovima. Opisani su slučajevi degenerativnih promjena u ćelijama korteksa prednjeg centralnog girusa, prednjih rogova kičmene moždine i malog mozga.
Kliničke manifestacije. Razvoj bolesti je postepen. Najčešće se prvi simptomi javljaju u drugoj deceniji života, iako postoje velike varijacije u dobi u kojoj bolest počinje. U početku se javlja ukočenost u nogama i brzi zamor pri hodanju, koji se povećava kako bolest napreduje. Razvija se karakterističan spastični hod, varusni i ekvinovarusni deformiteti stopala, promjene na stopalima tipa “Friedreichovo stopalo”, kontrakture tetiva i mišića, posebno kod skočni zglobovi. Postupno se povećava slabost u donjim ekstremitetima, ali se ne opaža potpuna paraliza donjih ekstremiteta. Prilikom kliničkog pregleda pacijenata, već u početnim stadijumima bolesti, otkriva se povećanje tetivnih refleksa, patološki refleksi grupa fleksije i ekstenzije (Babinsky, Oppenheim, Rossolimo, Gordon, Schaeffer, Bekhterev-Mendel, Zhukovsky), klonus stopala i koljena pojavljuju se rano. U većini slučajeva, kožni refleksi su očuvani, funkcije zdjeličnih organa nisu narušene. Nema poremećaja osjetljivosti. Inteligencija očuvana. Mnogo kasnije, gornji udovi su uključeni u patološki proces. Često je donja spastična parapareza praćena simptomima oštećenja vidnog i okulomotornog živca, nistagmusom, dizartrijom, ataksijom i intencionim tremorom.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza obično ne izaziva poteškoće ako postoji porodična priroda bolesti i tipična klinička slika.
U atipičnim sporadičnim slučajevima, bolest treba razlikovati od spinalnog oblika multipla skleroza, amiotrofična lateralna skleroza, tumori kičmene moždine i drugi patološki procesi različite etiologije, uzrokujući kompresiju kičmene moždine, kao i mijelozu uspinjača, neurosifilis i druge oblike cerebelarno-piramidalne degeneracije. Spinalni oblik multiple skleroze, uz donju spastičnu paraparezu, karakterizira recidivirajući tok, nestabilnost i privremena reverzibilnost pojedinih simptoma, disfunkcija zdjeličnih organa, gubitak ili asimetrija abdominalnih refleksa i asimetrija simptoma lezije općenito, promjene imunoloških parametara krvi i cerebrospinalne tekućine. Podaci o nasljednoj prirodi bolesti su od odlučujućeg značaja. Za razliku od amiotrofične lateralne skleroze, Strumpellova bolest počinje u u mladosti, nema znakova oštećenja perifernih motornih neurona (fascikularni trzaji, atrofija malih mišića šake, karakteristične EMG promjene), niti bulbarnih poremećaja. Kod diferencijacije od ekstramedularnih tumora i sindroma kompresije kičmene moždine druge etiologije, karakteristični su za tumore poremećaji segmentne osjetljivosti, asimetrija oštećenja ekstremiteta, prisustvo bloka subarahnoidalnog prostora i disocijacija proteina u likvoru tokom lumbalne punkcije. bitan. Kod neurosifilisa, za razliku od Štrumpelove bolesti, postoji anamneza kožne manifestacije. Vodeći u kliničku sliku su simptomi oštećenja stražnjih moždina kičmene moždine, karakteristični poremećaji zjenica, utvrđuju se promjene u krvi i likvoru.
Diferencijalna dijagnoza porodične spastične paraplegije s drugim degenerativnim lezijama kičmene moždine ponekad može biti teška. Pomaže u prepoznavanju simptoma oštećenja drugih dijelova nervnog sistema (cerebelarni, oftalmološki, itd.).
Tok i prognoza. Tok bolesti je polako progresivan; maligniji tok se bilježi kada se javi u ranoj dobi. Sa kasnim razvojem bolesti, hipertenzija i hiperrefleksija prevladavaju nad motoričkim poremećajima. Prognoza za život je povoljna. Stepen invaliditeta zavisi od težine disfunkcije nervnog sistema.
Tretman. Simptomatično. Propisani lijekovi koji smanjuju mišićni tonus su midokalm, baklofen, izoprotan (skutamil), sredstva za smirenje: sibazon (seduxen), nozepam (tazepam), hlozepid (elenium). Indicirane su fizioterapeutske procedure i aplikacije parafina na mišiće donjih ekstremiteta. Prijavite se akupresura, refleksologija, fizioterapija po potrebi ortopedske mjere. Indikovani su kursevi opšteg jačanja: vitamini B, metabolički lekovi: piracetam (nootropil), piriditol (encefabol), aminalon, cerebrolizin, aminokiseline, ATP, kokarboksilaza, lekovi koji poboljšavaju mikrocirkulaciju.
24.2.2. Parkinsonova bolest
Bolest je prvi opisao engleski lekar Džejms Parkinson, koji ju je nazvao paralizom drhtanja. 1877. Jean Martin Charcot je dodao kliničke karakteristike bolesti. Bolest se javlja kod 60-140 na 100.000 stanovnika; njegova učestalost naglo raste s godinama. Prema statistikama, paraliza drhtanja javlja se kod 1% populacije mlađe od 60 godina i kod 5% starijih osoba. Muškarci nešto češće obolijevaju od žena.
Etiologija i patogeneza. Kliničke manifestacije paralize drhtanja i sindroma parkinsonizma nastaju kao posljedica akutnog i hronične infekcije nervnog sistema (ekonomski epidemijski encefalitis, krpeljni, virusni i druge vrste encefalitisa). Uzroci bolesti mogu biti cerebralna ateroskleroza, vaskularne bolesti mozga, tumori, traume nervnog sistema, dugotrajna upotreba fenotiazinskih lekova (aminazin, triftazin), derivata rauvolfije, metildope - parkinsonizam izazvan lekovima. Parkinsonizam se može razviti uz akutnu ili kroničnu intoksikaciju ugljičnim monoksidom i manganom. U nastanku akinetičko-rigidnog sindroma može igrati ulogu nasljedni poremećaj metabolizma kateholamina u mozgu ili inferiornost enzimskog sistema koji kontrolira ovaj metabolizam. Porodična priroda bolesti često se otkriva autosomno dominantnim tipom nasljeđivanja. Takvi slučajevi se nazivaju Parkinsonova bolest. Različiti egzo- i endogeni faktori (ateroskleroza, infekcije, intoksikacije, ozljede) doprinose ispoljavanju genetskih defekata u mehanizmima metabolizma kateholamina u subkortikalnim jezgrima i nastanku bolesti.
Glavna patogenetska veza paralize drhtanja i sindroma parkinsonizma je kršenje metabolizma kateholamina (dopamin, norepinefrin) u ekstrapiramidnom sistemu. Dopamin obavlja nezavisnu posredničku funkciju u provođenju motoričkih činova. Normalno, koncentracija dopamina u bazalnim ganglijama je mnogo puta veća od njegovog sadržaja u drugim strukturama nervnog sistema. Acetilholin je posrednik ekscitacije između striatuma, globus pallidusa i supstancije nigre. Dopamin je njegov antagonist, djeluje inhibitorno. Kada su oštećena supstancija nigra i globus pallidus, nivo dopamina u kaudatnom jezgru i putamenu se smanjuje, odnos dopamina i norepinefrina se narušava i dolazi do poremećaja ekstra funkcije. piramidalni sistem. Normalno, impulsi se moduliraju ka supresiji kaudatnog jezgra, putamena, supstancije nigre i stimulacije globus pallidusa. Kada je funkcija supstancije nigre isključena, dolazi do blokade impulsa koji dolaze iz ekstrapiramidalnih zona moždane kore i strijatuma do prednjih rogova kičmene moždine. U isto vrijeme, ćelije prednjih rogova primaju patološke impulse iz globus pallidusa i crne supstance. Kao rezultat toga, povećava se cirkulacija impulsa u sistemu alfa i gama motornih neurona kičmene moždine s prevladavanjem alfa aktivnosti, što dovodi do pojave palidalno-nigralne rigidnosti mišićnih vlakana i tremora - glavnih znakova parkinsonizma. .
Patomorfologija. Glavne patološke promjene kod parkinsonizma uočavaju se u supstanci nigra i globus pallidusu u vidu degenerativnih promjena i smrti. nervne celije. Na mjestu mrtvih ćelija pojavljuju se žarišta proliferacije glijalnih elemenata ili ostaju praznine.
Kliničke manifestacije. Basic klinički sindrom– akinetičko-rigidni ili hipertenzivno-hipokinetički. Paralizu drhtanja i parkinsonizam karakteriziraju hipo- i akinezija. Pojavljuje se osebujan fleksijski položaj: glava i trup su nagnuti prema naprijed, ruke su polusavijene u laktovima, zglobovima i falangealnim zglobovima, često čvrsto dovedeni na bočne površine prsa i trupa, noge su polusavijene u kolenskih zglobova. Zapaženi su loši izrazi lica. Brzina voljnih pokreta postupno se usporava s razvojem bolesti, ponekad potpuna nepokretnost može nastupiti prilično rano. Hod karakteriziraju mali koraci koji se miješaju. Često postoji tendencija nevoljnog trčanja naprijed (pogon). Ako gurnete pacijenta naprijed, on trči da izbjegne pad, kao da "sustiže svoj centar gravitacije". Često pritisak na prsa dovodi do trčanja unazad (retropulzija) ili u stranu (lateropulzija). Ovi pokreti se takođe primećuju kada pokušavate da sednete, ustanete ili zabacite glavu unazad. Često, s izraženim sindromom, položaji pacijenata podsjećaju na kataleptičke. Akineza i plastična hipertenzija posebno su izražene u mišićima lica, žvakanja i okcipitalnih mišića, mišići udova. Prilikom hodanja nema prijateljskih pokreta ruku (aheirokineza). Govor je tih, monoton, bez modulacije, sa tendencijom da bledi na kraju fraze.
Prilikom pasivnog pokreta udova uočava se svojevrsni otpor mišića zbog povećanja tonusa mišića antagonista, fenomena „zupčanika“ (stiče se utisak da zglobna površina sastoji se od kombinacije dva zupčanici). Povećanje tonusa mišića antagonista tokom pasivnih pokreta može se odrediti sljedećom tehnikom: ako podignete glavu osobe koja leži, a zatim naglo otpustite ruku, glava neće pasti na jastuk, već će pasti relativno glatko. Ponekad je glava blago podignuta kada ležite - fenomen "imaginarnog jastuka".
Tremor je karakterističan, iako nije obavezan, simptom sindroma parkinsonizma. Ovo je ritmično, pravilno, nevoljno drhtanje udova, mišića lica, glave, donja vilica, jezik, izraženiji u mirovanju, smanjuje se pri aktivnim pokretima. Frekvencija oscilacija je 4-8 u sekundi. Ponekad se primjećuju pokreti prstiju u obliku "kotrljajućih pilula" ili "brojanja novčića". Tremor se pojačava od uzbuđenja i praktično nestaje u snu.
Mentalni poremećaji manifestiraju se gubitkom inicijative, aktivnosti, sužavanjem horizonata i interesa, naglim smanjenjem raznih emocionalne reakcije i afekti, kao i neka površnost i sporost razmišljanja (bradifrenija). Uočava se bradipsihija - teško aktivno prebacivanje s jedne misli na drugu, akairija - ljepljivost, viskoznost, egocentrizam. Ponekad se javljaju paroksizmi mentalnog uzbuđenja.
Autonomni poremećaji se manifestuju u vidu masnoće kože lica i tjemena, seboreje, hipersalivacije, hiperhidroze, trofičkih poremećaja u distalnim dijelovima ekstremiteta. Otkriva se kršenje posturalnih refleksa. Ponekad posebne metode Studija otkriva disanje koje je nepravilno po učestalosti i dubini. Tetivni refleksi su obično bez odstupanja. Kod aterosklerotskog i postencefalitičnog parkinsonizma mogu se otkriti pojačani refleksi tetiva i drugi znaci piramidalne insuficijencije. Kod postencefalitičnog parkinsonizma javljaju se takozvane okulogične krize - fiksacija pogleda prema gore nekoliko minuta ili sati; ponekad je glava zabačena unazad. Krize se mogu kombinovati sa kršenjem konvergencije i akomodacije (progresivna supranuklearna paraliza).
Uobičajeno je razlikovati nekoliko kliničkih oblika paralize drhtanja i parkinsonizma; kruto-bradikinetičko, drhtavo-kruto i drhtavo. Kruti-bradikinetički oblik karakterizira povećanje mišićnog tonusa prema plastičnom tipu, progresivno usporavanje aktivnih pokreta do nepokretnosti; javljaju se mišićne kontrakture i držanje fleksora kod pacijenata. Ovaj oblik parkinsonizma, najnepovoljniji po svom toku, češće se opaža kod aterosklerotskog, a rjeđe kod postencefalitičnog parkinsonizma. Drhtavo-rigidni oblik karakterizira drhtanje ekstremiteta, uglavnom njihovih distalnih dijelova, što je praćeno ukočenošću voljnih pokreta s razvojem bolesti. Drhtavi oblik parkinsonizma karakterizira prisustvo stalnog ili gotovo konstantnog drhtanja udova, jezika, glave i donje vilice srednje i velike amplitude. Tonus mišića je normalan ili blago povišen. Tempo voljnih pokreta se održava. Ovaj oblik je češći kod postencefalitičnog i posttraumatskog parkinsonizma.
Laboratorija i funkcionalne studije. Kod posttraumatskog parkinsonizma detektira se povećanje pritiska cerebrospinalne tekućine uz normalan ćelijski i proteinski sastav. Kod parkinsonizma, koji nastaje kao posljedica trovanja ugljičnim monoksidom, karboksihemoglobin se nalazi u krvi, a kod manganskog parkinsonizma tragovi mangana se nalaze u krvi, urinu i likvoru. Globalna elektromiografija kod paralize drhtanja i parkinsonizma otkriva kršenje mišićne elektrogeneze - povećanje bioelektrične aktivnosti mišića u mirovanju i prisutnost ritmičkih grupnih potencijalnih pražnjenja. Elektroencefalografija otkriva pretežno difuzne, grube promjene u bioelektričnoj aktivnosti mozga.
Dijagnoza i diferencijalna dijagnoza. Prije svega, potrebno je razlikovati Parkinsonovu bolest od sindroma parkinsonizma. Postencefalitički parkinsonizam karakteriziraju okulomotorni simptomi; Mogu se uočiti fenomeni tortikolisa i torzijske distonije, koji se nikada ne primjećuju kod paralize drhtanja. Javljaju se poremećaji spavanja, respiratorne diskinezije sa napadima zijevanja, kašlja, adiposogenitalni poremećaji i vegetativni paroksizmi. Posttraumatski parkinsonizam može se pouzdano dijagnosticirati kod mladih i sredovečnih pacijenata. Bolest se razvija nakon teške, ponekad ponovljene traumatske ozljede mozga. Posttraumatski parkinsonizam ne karakterišu antetropulzije, konvulzije pogleda, poremećaji žvakanja, gutanja, disanja i kataleptoidni fenomeni. Istovremeno, česti su vestibularni poremećaji, oštećenje inteligencije i pamćenja, te vizualne halucinacije (zbog oštećenja moždane kore). Često se opaža regresivni tok ili stabilizacija patološkog procesa. Za dijagnozu manganskog parkinsonizma, anamnezu (podaci o radu u kontaktu sa manganom ili njegovim oksidima), otkrivanje mangana u biološke tečnosti. Dijagnoza oksikarbonskog parkinsonizma zasniva se na određivanju karboksihemoglobina u krvi.
Kod aterosklerotskog parkinsonizma, drhtanje i ukočenost se kombinuju sa znacima cerebralne ateroskleroze ili se javljaju nakon akutnih poremećaja cerebralnu cirkulaciju. Focal neurološki simptomi u obliku piramidalne insuficijencije, izraženih pseudobulbarnih simptoma. Često se utvrđuje jednostranost krutosti i krutosti. U krvi se otkriva dislipidemija, karakteristična za aterosklerozu. Određene promjene u REG-u se bilježe u obliku spljoštenja pulsnih valova.
Kod senila se može uočiti klinička slika koja podsjeća na Parkinsonovu bolest aterosklerotska demencija, koji se najviše odlikuje grubošću mentalnih poremećaja sve do demencije. Rigidnost i ukočenost su umjereni, tremor obično izostaje. Pojedinačne kliničke manifestacije parkinsonizma mogu se otkriti i kod drugih nasljednih degenerativnih bolesti nervnog sistema: Friedreichova ataksija, olivopontocerebelarna atrofija, ortostatska hipokinezija, Creutzfeldt-Jakobova bolest. Kod ovih bolesti, uz akinetičko-rigidne simptome, javljaju se i progresivni fenomeni cerebelarne ataksije.
Tok i prognoza. Bolest stalno napreduje. Izuzetak su neki oblici uzrokovani intoksikacijom lijekovima (ako se lijekovi prekinu, stanje se može poboljšati). Općenito je prihvaćeno da liječenje u početna faza omogućava vam da smanjite ozbiljnost simptoma i usporite napredovanje bolesti. U kasnijim fazama, mjere liječenja su manje efikasne. Bolest dovodi do invaliditeta za nekoliko godina. Čak i liječenje levodopom trenutno usporava progresiju na kratak vremenski period. Ovo potvrđuje stav da u osnovi bolesti nije samo primarni biohemijski defekt, već i još neistraženi neuropatološki proces.
Tretman. Liječenje bolesnika s paralizom drhtanja i sindromom parkinsonizma treba biti kompleksno, dugotrajno i uključivati specifične antiparkinsonike, sedative, fizioterapeutske postupke, fizikalnu terapiju, psihoterapiju, uzimajući u obzir etiološki faktor, starost pacijenata, klinički oblik i stadijum bolesti, kao i prisustvo prateće bolesti. Za blage forme prvo se propisuju amantadin (midantan) i parasimpatolitici, jer manje uzrokuju nuspojave. Koriste se centralni parasimpatolitici (ciklodol, narkopan), piridoksin, amantadin, agonisti dopaminskih receptora (bromokriptin, lizurid).
Za teške kliničke manifestacije parkinsonizma, levodopa je trenutno osnovni lijek, obično u kombinaciji s inhibitorom dekarboksilaze. Doze se polako povećavaju tokom nekoliko sedmica dok se ne postigne klinički efekat. Nuspojave lijek – distonični poremećaji i psihoze. Levodopa, ulazeći u centralni nervni sistem, dekarboksilira se u dopamin, koji je neophodan za normalnu funkciju bazalnih ganglija. Lijek djeluje prvenstveno na akineziju i, u manjoj mjeri, na druge simptome. Kada se levodopa kombinuje sa inhibitorom dekarboksilaze, doza levodope se može smanjiti i time smanjiti rizik od nuspojava.
U arsenalu simptomatskih antiparkinsonika veliko mjesto zauzimaju antiholinergici, koji blokiranjem m- i n-holinergičkih receptora pospješuju opuštanje prugasto-prugastih i glatkih mišića, smanjuju nasilne pokrete i pojave bradikinezije. To su prirodni i sintetički lijekovi slični atropinu: bellazone (romparkin), norakin, combipark. Koriste se i fenotiazinski lijekovi: dinezin, deparkol, parsidol, diprazin. Glavni razlog za raznolikost lijekovi, koristi se za liječenje parkinsonizma, nedovoljno terapeutska efikasnost, prisutnost nuspojava, individualna netolerancija i brza ovisnost o njima.
Operacija. Uprkos velikim uspesima postignutim u liječenje lijekovima parkinsonizma, njegove mogućnosti su u nekim slučajevima ograničene.
Najrasprostranjeniji lijek, levodopa, u većoj mjeri pomaže u otklanjanju simptoma bolesti kao što su akinezija i opća ukočenost, au manjoj mjeri utiče na rigidnost mišića i tremor. U otprilike 25% pacijenata ovaj lijek je praktički nedjelotvoran ili se loše podnosi.
U tim slučajevima postoje indikacije za stereotaktičku operaciju na subkortikalnim čvorovima. Obično se vrši lokalna destrukcija ventrolateralnog jezgra thalamus optis, subtalamičkih struktura ili globus pallidusa.
Uz pomoć operacije u većini slučajeva moguće je postići pozitivan učinak - smanjenje mišićnog tonusa, slabljenje ili prestanak tremora, smanjenje hipokinezije.
Operacija se obično izvodi na strani suprotnoj od one na kojoj prevladavaju simptomi Parkinsonove bolesti. Kada je indicirano, vrši se bilateralna destrukcija subkortikalnih struktura.
IN poslednjih godina Za liječenje parkinsonizma također se koristi implantacija embrionalnog nadbubrežnog tkiva u striatum. Prerano je govoriti o kliničkoj efikasnosti ovakvih operacija.
Stereotaktičke operacije na subkortikalnim strukturama koriste se i kod drugih bolesti praćenih nasilnim pokretima (hemibalizam, koreoatetoza, tortikolis i neke druge).
Radna sposobnost kod Parkinsonove bolesti i parkinsonizma zavisi od težine motoričkih poremećaja, vrste profesionalna aktivnost. Za pluća i umjereni prekršaji motoričke funkcije, pacijenti ostaju sposobni za rad dugo vremena razne vrste mentalni rad, kao i rad koji nije povezan sa fizičkim stresom i izvođenjem preciznih i koordinisanih pokreta. Uz teške manifestacije bolesti, pacijenti su nesposobni za rad i potrebna im je vanjska pomoć.
24.2.3. Hepatocerebralna distrofija
Hepatocerebralna distrofija (hepatolentikularna degeneracija, Westphal-Wilson-Konovalov bolest) je kronična progresivna nasljedna degenerativna bolest koju karakterizira kombinirano oštećenje subkortikalnih čvorova centralnog nervnog sistema i jetre. Opisao 1883. K. Westphal i 1912. S. Wilson. Termin "hepatocerebralna distrofija" predložio je N.V. Konovalov.
Etiologija i patogeneza. Bolest je nasledna. Tip nasljeđivanja je autosomno recesivan. Vodeća patogenetska karika je genetski determinisani poremećaj u sintezi proteina ceruloplazmina, koji je dio a2-globulina koji prenosi bakar. Kao rezultat, stvara se visoka koncentracija bakra u krvi i njegovo taloženje se dešava u organima i tkivima, uglavnom u jetri, mozgu, rožnici, kao i u bubrezima i drugim organima. Toksičan efekat bakar je povezan sa blokom sulfhidrilnih grupa u oksidativnim enzimima, što dovodi do poremećaja redoks procesa u ćeliji.
Patomorfologija. U mozgu, jetri, bubrezima, slezeni, rožnici, šarenici i očnom sočivu otkrivaju se degenerativne promjene, najizraženije u subkortikalnim jezgrama. Otkrivaju se i distrofične promjene u nervnim stanicama, žarišno omekšavanje moždanog tkiva sa stvaranjem mikrocista i proliferacija glije. Otkrivaju se promjene u malim žilama moždanog tkiva, krvarenja oko njih i perivaskularni edem. Stalni znak Bolest je ciroza jetre.
Kliničke manifestacije. Sastoje se od simptoma oštećenja centralnog nervnog sistema i unutrašnjih organa. Pacijenti se razvijaju i povećavaju rigidnost mišića, razne hiperkineze, pseudobulbarni simptomi, progresivni pad inteligencije, disfunkcija jetre i promjene na šarenici. Vodeći je sindrom ekstrapiramidnih poremećaja: ukočenost mišića trupa, udova, lica, ždrijela i, kao posljedica toga, poremećaji u hodu, gutanju i govoru. Istovremeno dolazi do hiperkineze različite prirode: tremor, atetoza, torziona distonija, namjerni tremor, pojačan pri pokušaju izvođenja voljnih pokreta. Hiperkineza je nepravilne prirode.
Ovisno o težini i kombinaciji kliničkih manifestacija, dobi u kojoj se bolest pojavila i stupnju oštećenja jetre razlikuju se četiri oblika hepatocerebralne distrofije.
1. Rani rigidno-aritmohiperkinetički oblik ima najmaligniji tok. Neurološke manifestacije se razvijaju u dobi od 7 do 15 godina. Tome obično prethode znaci oštećenja jetre. Kliničkom slikom dominiraju rigidnost mišića i hiperkineza.
2. Drhtavo-rigidni i drhtavi oblici, koji se javljaju u kasnijoj dobi (17-20 godina). Karakterizira ga istovremena pojava ukočenosti i drhtanja, što je često prvi znak bolesti; postepeno se povećava, može postati opći, zahvaćajući mišiće trupa, udova, lica, čeljusti, mekog nepca, epiglotisa, glasnih žica, respiratornih mišića i dijafragme. Gutanje je otežano, govor postaje pevan. Često se primjećuju izražene mentalne promjene.
3. Ekstrapiramidno-kortikalni oblik, identifikovan od strane N.V. Konovalov, odlikuje se poremećajem višeg funkcije mozga, prisustvo paralize, često epileptični napadi, ozbiljno smanjenje inteligencije s promjenama ličnosti.
4. Abdominalni oblik karakteriše pretežno oštećena funkcija jetre. Neurološki simptomi se javljaju u kasnijim stadijumima bolesti.
Tok i prognoza. Kurs stalno napreduje. Očekivano trajanje života ovisi o kliničkom obliku bolesti i pravovremenosti započetog liječenja. Prosječan životni vijek pacijenata bez liječenja je oko 6 godina.
Podaci iz laboratorijskih i funkcionalnih studija. U krvnom serumu detektuje se značajno smanjenje sadržaja ceruloplazmina (ispod 10 IU pri normi od 25-45 IU), hipoproteinemija, hiperkupurija (do 1000 mcg/dan i više pri normi od 150 mcg/dan) i hiperaminoacidurija (do 1000 mg/dan pri normi od 350 mg/dan). Moguće je i povećanje nivoa amonijaka u krvi i promene u testovima jetre. Patognomonični znak Hepatocerebralna distrofija je Kayser-Fleischer prsten rožnice, koji je taloženje pigmenta koji sadrži bakar duž periferije šarenice.
Diferencijalna dijagnoza. Bolest treba razlikovati od letargičnog encefalitisa, horee minor, degenerativnih subkortikalnih bolesti i multiple skleroze. Identifikacija porodične anamneze, karakteristična klinička slika, Kayser-Fleischer kornealni prsten, nizak nivo ceruloplazmina u krvi i povećano izlučivanje bakra u urinu kod pacijenata i njihovih srodnika omogućava nam postavljanje dijagnoze hepatocerebralne distrofije.
Tretman. Glavni cilj tretmana je uklanjanje viška bakra iz organizma. U tu svrhu koriste se tiolni lijekovi, koji uključuju unitiol, dekaptol i D-penicilamin. Doze se biraju pojedinačno. O-penicilamin se propisuje u prosjeku u dozi od 0,45 do 2 g dnevno nakon jela. Lijek se mora uzimati cijeli život.
Liječenje je najefikasnije u ranim stadijumima bolesti. Unithiol se propisuje u ponovljenim kursevima od 5 ml 5% rastvora intramuskularno dnevno ili svaki drugi dan (kurs od 25 injekcija sa pauzom između kurseva od 5-6 meseci). Liječenje se kombinira s lijekovima koji poboljšavaju funkciju jetre. Preporučeno posebna dijeta uz ograničavanje namirnica bogatih bakrom (jetra, pečurke, čokolada, ostrige itd.), životinjskih masti, proteina. Hrana treba da bude bogata vitaminima i ugljenim hidratima.
24.2.4. Torziona distopija
Kronična progresivna bolest nervnog sistema koja se klinički manifestira promjenama mišićnog tonusa i nevoljnim toničkim kontrakcijama mišića trupa i udova.
Etiologija i patogeneza. Postoje idiopatska (porodična) torzija i simptomatska distonija. Tip nasljeđivanja idiopatske torzijske distonije je i autosomno dominantan i autosomno recesivan. Simptomatska torziona distonija javlja se kod hepatocerebralne distrofije, Huntingtonove horeje, tumora mozga, epidemijskog encefalitisa, djetinjstva cerebralna paraliza. Postoje indicije da je poremećaj metabolizma dopamina važan u patogenezi nasljedne torzijske distonije. Prilikom pregleda ovih pacijenata, uočeno je povećanje sadržaja dopamin-β-hidroksilaze u krvnom serumu.
Patomorfologija. Distrofične promjene nalaze se uglavnom u malim neuronima u predjelu ljuske lentiformnog jezgra, rjeđe u drugim bazalnim ganglijima.
Kliničke manifestacije. Bolest se razvija postepeno, u 2/3 slučajeva mlađih od 15 godina. U djetinjstvu prvi simptomi bolesti mogu biti poremećaj hoda, spastični tortikolis; Kod odraslih, primarni generalizirani oblici su češći. Kao rezultat kršenja omjera funkcija sinergističkih i antagonističkih mišića, javljaju se nasilne dugotrajne tonične kontrakcije mišića trupa, glave, zdjeličnog pojasa i udova, obično rotacijske prirode, u kombinaciji s atetoidnim pokretima. u prstima. Čini se da se mišići stalno kontrahuju kako bi savladali djelovanje antagonista. Nastali položaji, čak i oni najneudobniji, traju dugo vremena. Hiperkineza se pojačava uzbuđenjem, aktivnim pokretima i nestaje tokom spavanja. Postepeno, kako bolest napreduje, pacijentovo držanje postaje konstantno distonično, uz povećanje lumbalna lordoza, fleksija kuka, medijalna rotacija ruku i nogu. Ovisno o učestalosti distoničnih pojava, razlikuju se lokalni i generalizirani oblici bolesti. Kod lokalnih distoničnih simptoma dolazi do tonične kontrakcije pojedinih mišićnih grupa, poremećeni su voljni pokreti i dolazi do abnormalnog držanja. Takvi simptomi uključuju spazmodični tortikolis, pisčev grč, oromandabularnu distoniju (otvaranje i zatvaranje usta i nevoljni pokreti jezika), blefarospazam, bukofacijalnu, bukolingvalnu distoniju, koreoatetozu.
Tok i prognoza. Bolest u većini slučajeva napreduje stabilno. Ponekad postoje različita trajanja remisije. Brzo nastupa duboka invalidnost pacijenata i smrt, posebno u generaliziranom obliku.
Tretman. Dugotrajno, simptomatično. Koriste se kombinacije antiholinergika i sedativa; u nekim slučajevima je efikasna upotreba levodope. Takođe se propisuje haloperidol ili rezerpin. Vrlo rijetko pribjegavaju stereotaktičkim operacijama na subkortikalnim jezgrima.
24.2.5. Huntingtonova koreja
Kronična progresivna nasljedna degenerativna bolest koju karakterizira pojačana horeička hiperkineza i demencija. Opisao J. Huntington 1872. Učestalost Huntingtonove horeje je od 2 do 7 slučajeva na 100.000 stanovnika. Takođe se koristi i termin „horička demencija“.
Etiologija i patogeneza. Bolest je nasledna. Tip nasljeđivanja je autosomno dominantan sa visokom penetracijom (80-85%). Muškarci češće obolijevaju. Patogeneza nije dovoljno proučavana. U nekim slučajevima utvrđen je nedostatak GABA u moždanim stanicama, povećanje sadržaja željeza u stanicama supstancije nigre, te poremećaji u metabolizmu dopamina. Proučavan je patofiziološki mehanizam poremećaja kretanja. Blok strionigralnih veza uzrokuje nedostatak kontrole nad koordinacijom pokreta i tonusom mišića od strane supstancije nigre, koja prenosi impulse primljene iz premotorne zone korteksa do ćelija prednjih rogova kičmene moždine u nepravilan niz.
Patomorfologija. Otkriva se atrofija mozga. U subkortikalnim ganglijima, uglavnom u putamenu i kaudatnom jezgru, otkrivaju se grube degenerativne promjene u malim i velikim stanicama, smanjenje njihovog broja i proliferacija glijalnih elemenata.
Kliničke manifestacije. Bolest se obično javlja u dobi od 30 godina i više. Prvi simptomi mogu biti intelektualni poremećaji, a zatim se postepeno razvija demencija. Istovremeno se javlja koreična hiperkineza: brzi, nepravilni, nepravilni pokreti u različitim mišićnim grupama. Izvođenje voljnih pokreta je otežano zbog hiperkineze i praćeno je brojnim nepotrebnim pokretima. Na primjer, prilikom hodanja pacijenti prave grimasu, gestikuliraju, čučnu i široko rašire ruke. Međutim, čak i uz izraženu hiperkinezu, posebno na početku bolesti, mogu je svjesno potisnuti. Govor je otežan i takođe praćen pretjeranim pokretima. Tonus mišića je smanjen. Pareza udova i drugi fokalni neurološki simptomi nisu otkriveni. Često se primjećuju endokrini i neurotrofični poremećaji. U 5-16% slučajeva dijagnosticira se atipična akinetičko-rigidna varijanta Huntingtonove koreje. U ovom slučaju, akinetičko-rigidni sindrom se razvija u kombinaciji s progresivnom intelektualnom degradacijom i umjerenom koreičnom hiperkinezom. Od nasilnih pokreta prevladava koreoatetoza.
Protok. Bolest stalno napreduje. Njegovo trajanje je 5-10 godina od trenutka pojave prvih simptoma. U atipičnom akinetičko-rigidnom obliku opaža se benigniji tok.
Podaci iz laboratorijskih i funkcionalnih studija. EEG pokazuje difuzne promene bioelektrična aktivnost mozga. Kompjuterizirana tomografija i magnetna rezonanca otkrivaju dilataciju ventrikula i takozvanu depresiju talamusa; ako je bolest povezana s oštećenjem njegovih malih stanica, otkrivaju se znakovi atrofije moždane kore. Postoje indicije o mogućnosti rane, pretkliničke dijagnoze bolesti na osnovu proučavanja osjetljivosti limfocita krvi na rendgensko zračenje.
Dijagnoza i diferencijalna dijagnoza. Dijagnoza može biti teška u atipičnim slučajevima Huntingtonove koreje.
U svim slučajevima veliki značaj imaju porodičnu prirodu bolesti, identifikujući druge žarišne simptome oštećenje mozga, priroda bolesti, promjene u likvoru i drugi dijagnostički kriteriji.
Huntingtonovu koreju treba razlikovati od horeičnog sindroma koji se javlja kod tumora mozga, od sifilisa, encefalitisa, vaskularne bolesti, kao i senilna (senilna) koreja.
Tretman. Za suzbijanje hiperkineze propisuju se antagonisti dopamina. To su lijekovi iz serije fenotiazina - triftazin (7,5-10 mg dnevno) u kombinaciji sa sredstvima za smirenje, dopegitom, rezerpinom. Pokušaji liječenja pacijenata koji pate od Huntingtonove horeje stereotaktičkim operacijama bili su neuspješni.
24.2.6. Friedreichova bolest
Friedreichova porodična ataksija je nasledna degenerativna bolest nervnog sistema, koju karakteriše sindrom oštećenja zadnje i bočne moždine kičmene moždine. Tip nasljeđivanja je autosomno recesivan, sa nepotpunom penetracijom patološkog gena. Muškarci i žene podjednako često obolijevaju.
Patomorfologija. Degenerativne promjene nalaze se u provodnim putevima zadnje i lateralne moždine kičmene moždine, uglavnom u Gaulleovim snopićima, u manjoj mjeri – Burdach, Flexig, Gowers, vlaknima piramidalnog trakta, dorzalnim korijenima, kao i u ćelije malog korteksa, subkortikalnih ganglija i moždane kore.
Kliničke manifestacije. Početak bolesti datira od 6-15 godina života. Prvi simptom bolesti je nesiguran hod, koji je Charcot okarakterisao kao tabetično-cerebelarni. U ranim fazama, ataksija je izražena uglavnom na nogama. Kako bolest napreduje, problemi s koordinacijom se šire na gornje udove i lice. Neurološki pregled otkriva veliki nistagmus, ataksiju u rukama i nogama, adijadohokinezu, dismetriju, skenirani govor, poremećaje mišićno-zglobnog čula i osjetljivosti na vibracije. Promjene u rukopisu. Rani simptom je smanjenje, a zatim i nestanak tetivnih i periostalnih refleksa. Tonus mišića je smanjen. U kasnijim stadijumima bolesti javlja se aferentna pareza donjih, a zatim gornjih ekstremiteta, česti su patološki piramidalni refleksi i atrofija distalnih mišića. Inteligencija je smanjena.
Bolest napreduje sporo. Prosječan životni vijek je 10-15 godina od trenutka njegovog razvoja.
Dijagnoza i diferencijalna dijagnoza. Bolest se prepoznaje na osnovu karakteristični simptomi– deformiteti stopala tipa Friedreich (visoki svod, ekstenzija glavnih falangi prstiju i fleksija terminalnih falanga), oštećenje miokarda, endokrini poremećaji.
Bolest treba razlikovati od cerebralnog sifilisa, multiple skleroze, funikularne mijeloze i drugih oblika cerebelarne degeneracije.
Tretman. Koriste se simptomatski lijekovi: restorativni lijekovi, fizikalna terapija, masaža. U nekim slučajevima se to radi hirurška korekcija deformiteti stopala
24.2.7. Nasljedna cerebelarna ataksija Pierre Marie
Cerebelarna ataksija Pierre Marie je nasljedna degenerativna bolest koja prvenstveno zahvaća mali mozak i njegove puteve. Tip nasljeđivanja je autosomno dominantan. Bolest se javlja u dobi od 20 godina i više.
Patomorfologija. Otkriva se degenerativno oštećenje ćelija korteksa i jezgara malog mozga, spinocerebelarnih puteva u bočnim moždinama kičmene moždine, u jezgrima mosta i oblongate moždine.
Kliničke manifestacije. Bolest se manifestuje disfunkcijom malog mozga i njegovih veza. Ataksija se opaža prilikom izvođenja testova koordinacije, poremećaja hoda, skeniranog govora, tremora namjere i nistagmusa. Cerebelarni simptomi kombiniraju se s umjerenim ili teškim znacima piramidalne insuficijencije (pojačani tetivni i periostalni refleksi, klonus stopala), a ponekad i s okulomotornim poremećajima (strabizam, ptoza, insuficijencija konvergencije). Karakteristična karakteristika je izraženo smanjenje inteligencije u različitom stepenu.
Dijagnoza i diferencijalna dijagnoza. Najveće poteškoće nastaju u razlikovanju nasljedne cerebelarne ataksije Pierre Marie i Friedreichove ataksije. Potrebno je uzeti u obzir vrstu nasljeđivanja bolesti, dob u kojoj se javljaju prvi simptomi, prirodu promjena tetivnih refleksa (kod Friedreichove ataksije oni su smanjeni), prisutnost vidnih i okulomotornih poremećaja kod Pierre Mariea. ataksija, deformiteti stopala i skeleta. Multipla skleroza za razliku od porodična ataksija Pierre Marie karakterizira remitentni tok, veća težina donje spastične parapareze i disfunkcija karličnih organa.
Tretman. Simptomatično.
24.2.8. Olivopontocerebelarne degeneracije
Grupa nasljednih bolesti nervnog sistema, karakterizirana degenerativnim promjenama u neuronima malog mozga, jezgri donjih maslina i mosta, u nekim slučajevima - jezgri kranijalnih nerava kaudalne grupe, au manjem broju stepen – oštećenje puteva i ćelija prednjih rogova kičmene moždine, bazalnih ganglija. Bolesti se razlikuju po vrsti nasljeđivanja i različitim kombinacijama kliničkih simptoma. Prema klasifikaciji Koenigsmarka i Weinera, postoji 5 tipova olivopontocerebelarne degeneracije.
Tip I – Mendelijska olivopontocerebelarna degeneracija.Nasljeđuje se autosomno dominantno. Kurs je polako progresivan. Može se pojaviti u dobi od 11 do 60 godina. Kliničku sliku čine simptomi oštećenja malog mozga (ataksija, mišićna hipotonija, skenirani govor sa elementima dizartrije, intencioni tremor), jezgra kaudalnih kranijalnih nerava (dizartrija, disfagija), subkortikalnih ganglija (hiperkineza); piramidalni i okulomotorni simptomi su rjeđi.
Tip II – olivopontocerebelarna degeneracija Fickler-Winkler.Nasljeđuje se autosomno recesivno. Manifestuje se u dobi između 20 i 80 godina sa simptomima oštećenja malog mozga, pretežno ataksijom u udovima. Osjetljivost i tetivni refleksi su nepromijenjeni. Nema pareza.
Tip III – olivopontocerebelarna degeneracija sa degeneracijom retine.Nasljeđuje se autosomno dominantno. Javlja se u mladosti. Uz cerebelarne i ekstrapiramidne simptome utvrđuje se progresivno smanjenje vidne oštrine zbog pigmentne degeneracije ganglijskih stanica retine.
Tip IV – olivopontocerebelarna degeneracija Shute-Hyckmana.Nasljeđuje se autosomno dominantno. Manifestuje se u detinjstvu i adolescenciji. Pored cerebelarnih simptoma, otkriva se i oštećenje jezgara VII, IX, X i XII para kranijalnih nerava (paraliza facijalnog živca, bulbarni simptomi) i stražnje moždine kičmene moždine (poremećaji mišićno-zglobnog osjeta i osjetljivosti na vibracije).
Tip V – olivopontocerebelarna degeneracija sa demencijom, oftalmoplegijom i ekstrapiramidnim poremećajima. Tip nasljeđivanja je autosomno dominantan. Razvija se u srednjim godinama. Karakteriziraju ga demencija, progresivna oftalmoplegija, ekstrapiramidni i cerebelarni simptomi.
Olivopontocerebelarne degeneracije treba razlikovati od nasljedne Friedreichove i Pierre Marie ataksije, progresivnih oblika multiple skleroze, tumora malog mozga i juvenilnih oblika parkinsonizma.
Tretman. Simptomatično. Sprovode kurseve nespecifičnog restorativnog tretmana, masaže i fizikalne terapije.
Pošaljite svoj dobar rad u bazu znanja je jednostavno. Koristite obrazac ispod
Studenti, postdiplomci, mladi naučnici koji koriste bazu znanja u svom studiranju i radu biće vam veoma zahvalni.
Objavljeno na http://www.allbest.ru/
Urođena patologija nervnog sistema kod dece
Uvod
Simptomi poremećaja
Hidrocefalus
Sindrom poremećaja kretanja
Pareza i paraliza
Miatonični sindrom
Konvulzivni sindrom
Genetske lezije N.s.
Daunova bolest
Zaključak
Književnost
Uvod
Ne postoji nijedna bolest u kojoj centralni nervni sistem nije u nekoj mjeri uključen. Kod bilo kojeg patološkog procesa dolazi do iritacije nervnih završetaka zahvaćenog područja. Ova iritacija se prenosi odgovarajućim nervnim provodnicima do nervnih centara i izaziva odgovarajuću reakciju nervnog sistema. U svom radu ću se dotaknuti kongenitalne bolesti nervnog sistema (u daljem tekstu n.s.) kod djece.
Simptomi poremećaja
Simptomi poremećaja n.s. može biti vrlo raznolika, što može otežati dijagnozu, posebno u djetinjstvu.
Od tako upečatljivih simptoma kao što su konvulzije, paraliza, senzorni poremećaji, visoki ili niski tetivni refleksi, mentalna retardacija do simptoma koji na prvi pogled nisu vezani za nervni sistem, kao što su enureza kod epilepsije ili kožni osip i svrab kod neurodermatitisa.
Oštećenje nervnog sistema kod novorođenčadi može nastati i u maternici (prenatalno) i tokom porođaja (intrapartum).
Ako štetni faktori djeluju na dijete u embrionalnoj fazi intrauterinog razvoja, nastaju teški, često nekompatibilni sa životom defekti.
Ako je štetno djelovalo na dijete nakon 28 sedmica intrauterinog razvoja, tada dijete neće imati nikakvih mana, ali se kod normalno formiranog djeteta može javiti bilo kakva bolest.
Razne akutne ili hronične bolesti majke, rad u opasnim hemijskim industrijama ili rad povezan sa raznim zračenjima, kao i loše navike roditelji - pušenje, alkoholizam, narkomanija.
Na dijete koje raste u maternici mogu negativno utjecati teška toksikoza trudnoće, patologija djetetovog mjesta - placente i prodiranje infekcije u maternicu.
Hipertenzivno-hidrokefalni sindrom
Prilikom pregleda bolesnog dojenčeta utvrđuje se ekspanzija ventrikularnog sustava mozga, otkriva ultrazvukom mozga i bilježi se povećanje intrakranijalnog tlaka (kao što pokazuje ehoencefalografija). Kod hipertenzivno-hidrocefaličnog sindroma može dominirati ili hidrocefalus, koji se manifestuje ekspanzijom ventrikularnog sistema mozga, usled prekomerne akumulacije cerebrospinalne tečnosti (CSF) u komorama mozga ili subarahnoidalnog prostora, što je praćeno njihovim širenjem, ili hipertenzijski sindrom sa povišenim intrakranijalnim pritiskom. Kada preovladava povišeni intrakranijalni pritisak, dijete je nemirno, lako uzbudljivo, razdražljivo, često glasno vrišti, lagano spava, a dijete se često budi. Kada prevladava hidrocefalični sindrom, djeca su neaktivna, letargija i pospanost, a ponekad se primjećuju i zastoji u razvoju.
Često, kada se intrakranijalni tlak poveća, djeca zamagljuju oči, povremeno se pojavljuje Graefeov simptom (bijela pruga između zjenice i gornji kapak), au teškim slučajevima može doći do simptoma „zalazećeg sunca“, kada je šarenica oka, poput sunca na zalasku, do pola uronjena ispod donjeg kapka; ponekad se pojavljuje konvergentni strabizam, beba često zabacuje glavu unazad. Tonus mišića može biti smanjen ili povećan, posebno u mišićima nogu, što se manifestuje time što se pri osloncu stoji na prstima, a pri pokušaju hoda prekriži noge. Progresija hidrocefaličnog sindroma manifestuje se povećanjem mišićnog tonusa, posebno u nogama, dok su refleksi podrške, automatsko hodanje i puzanje smanjeni.
U slučajevima teškog, progresivnog hidrocefalusa, može doći do napadaja.
Hidrocefalus
Hidrocefalus se može javiti sa povećanim intrakranijalnim pritiskom i sa normalnim intrakranijalnim pritiskom.
Uzrok kongenitalnog hidrocefalusa koji se razvija u maternici je u pravilu različit intrauterine infekcije- citomegalija, sifilis, toksoplazmoza, akutni virusne infekcije, rubeola, itd., kao i defekti u razvoju mozga. Tokom porođaja može doći do intraventrikularnog krvarenja, što može uzrokovati i hidrocefalus.
Uzrok stečenog hidrocefalusa, koji se razvija nakon rođenja, najčešće su upalne infekcije i bolesti mozga.
Sindrom poremećaja kretanja. Pareza i paraliza
Downov sindrom hidrocefalus paraliza
U slučajevima paralize ili pareze dolazi do smanjenja tonusa u mišićnim grupama (udovi, lice, ždrijelo, itd.) do potpunog nestanka. Paraliza može nastati zbog smrti ili oštećenja motornog neurona (centralnog ili perifernog) ili njegovih procesa.
Među parezama su:
monopareza - oštećenje samo jednog ekstremiteta;
hemipareza - oštećenje ruke i noge s jedne strane;
gornja parapareza - oštećenje obje ruke;
donja parapareza --
oštećenje obe noge;
tetrapareza --
oštećenje obe ruke
i obe noge.
Motorni poremećaji nastaju kada je oštećen motorni nervni put koji se sastoji od dva neurona - centralnog i perifernog.
Centralni motorni neuron nalazi se u moždanoj kori u motornoj zoni prednjeg centralnog girusa. Njegov dug proces prolazi kroz glavne duboke strukture mozga, uključujući moždano stablo, dok daje vlakna jezgrima kranijalnih nerava. Na granici produžene moždine i kičmene moždine, vlakna središnjeg motornog neurona prelaze na suprotnu stranu, ulaze u bočne moždine kičmene moždine i spuštaju se u svom sastavu do najnižih dijelova. Duž kičmene moždine, vlakna se protežu od motornog trakta do svakog segmenta, završavajući na ćelijama prednjih rogova kičmene moždine.
Oštećenje centralnog motornog neurona u bilo kom segmentu dovodi do trenutnog razvoja centralne „spastične“ pareze, koja ima veoma tipične znakove: povećan mišićni tonus (hipertonus), visoki refleksi tetiva (hiperrefleksija), patološki znaci stopala.
Drugi neuron motoričkog puta - periferni - su motoričke ćelije prednjih rogova, smještene kroz kičmenu moždinu od vrha do dna. Vlakna iz perifernih neurona izlaze kao dio prednjih korijena iz kičmene moždine i učestvuju u formiranju pleksusa iz kojih se miješaju perifernih nerava koji sadrže senzorne i motoričke filamente. Završava se drugi motorni neuron u mišićima. Oštećenje drugog neurona motoričkog puta na bilo kojem segmentu dovodi do razvoja periferne „mladave“ pareze ili paralize ekstremiteta, koju karakterizira smanjen tonus mišića (hipotonija), smanjeni tetivni refleksi (hiporefleksija), praćena „težinom gubitak” i atrofija mišića.
Manifestacija smanjenog mišićnog tonusa s mlohavim parezom u nogama je karakteristična "žablja poza" - noge se otvaraju i padaju u stranu. Iz istog razloga, uz parezu jedne noge, može se prepoznati simptom "padajućeg stopala", što ukazuje na slabost mišića ove noge. Kod mlohave pareze, uglavnom u donjim dijelovima obje noge (u stopalima), može se javiti simptom „petnih stopala“, kada ljekar može lako dodirnuti stražnji dio stopala novorođenčeta s prednjom površinom potkoljenice.
Naprotiv, kod spastične pareze šaka, šake koje su čvrsto stisnute u šaku su udarne, a ruke su savijene u lakatnog zgloba, a ponekad se pelenski osip pojavljuje čak i na pregibima dlanova. Zanimljiva je posebna "ukočenost" udova.
Miatonični sindrom
Miatonični sindrom je jedan od najčešćih kompleksa neuroloških simptoma u neurologiji novorođenčadi. S vremenom može potpuno nestati, ali kod neke djece posljedice traju cijeli život. Rezultat izrazitog smanjenja mišićnog tonusa kod djece u rastu je prekomjerna pokretljivost u svim zglobovima udova do čujnog krckanja, hiperekstenzija (recurvacija) u zglobovima lakta i koljena, kada ruka ili noga imaju oblik luk kada je produžen. Za takve pacijente tipičan je simptom "palca", kada se dijete može mirno sagnuti thumb ruke do podlaktice.
Obično imaju zastoj u motoričkom razvoju: takva djeca nešto kasnije počinju držati glavu podignutu, sjediti i hodati. Više vole da sjede, ali kada počnu hodati, često padaju i brzo se umaraju.
Prije svega žig miatonični sindrom iz drugih stanja u kojima je moguće smanjenje mišićnog tonusa - kombinacija ravnomjernog smanjenja mišićnog tonusa s visokim tetivnim refleksima koje je identificirao neurolog. Razlog tome je oštećenje motoričkog piramidalnog trakta na istom nivou moždanog stabla gdje se nalazi retikularna formacija.
Konvulzivni sindrom
"Epilepsija" je grčka riječ prevedena kao "uhvatiti" ili "napasti". Statistike pokazuju da oko 50 miliona ljudi na planeti pati od ove bolesti! Razlozi koji uništavaju normalno bioelektrična aktivnost mozga, formirajući epileptičku aktivnost, krše se odnosi između aktivne supstance, moždani medijatori inhibitorne i ekscitatorne prirode. Važni su i poremećaji permeabilnosti nervnih ćelija i metabolizma vode i soli.
Za sada, sve ovo patoloških procesa nastavljaju neprimjetno, ali počinju se pojavljivati s ozljedama mozga, cerebrovaskularnim nesrećama, tumorima, infekcijama i intoksikacijama. U nekim slučajevima za razvoj epilepsije dovoljni su nasljedni faktori i kongenitalna predispozicija.
Postoje lokalizirani i generalizirani oblici epilepsije. Lokalizirane forme nazivaju se i lokalnim, žarišnim ili parcijalnim. U takvim slučajevima epileptički žarište je lokalizirano u određenom području moždane kore. U generaliziranim oblicima, cijeli mozak učestvuje u procesu napadaja. Moguć je postupni razvoj epileptičkog procesa: počevši od lokalnog napada, napad se zatim transformira u generalizirani. Takvi epileptični napadi nazivaju se sekundarno generaliziranim.
Prva faza napadaja je tonična, koja se manifestuje napetošću u mišićima trupa i udova. U ovoj fazi disanje prestaje na 30-40 sekundi, lice postaje cijanotično. Tada se obnavlja disanje, napad prelazi u toničnu fazu. Ritmički trzaji počinju u rukama i nogama. Trzanje ruku i nogu su klonične konvulzije.
Konvulzije se povuku, a nakon njih se razvija postiktalni san. Zbog toničnog grča žvačnih mišića, pacijent može ugristi jezik. Na vrhuncu napadaja to je moguće nehotično mokrenje. Napadi se obično dobro završavaju.
Uočene su mnoge karakteristike u manifestaciji velikih generaliziranih napadaja kod djece
Konvulzije kod djeteta mogu biti samo tonički ili samo klonični. Dijete se „napinje, rasteže, postaje napeto, tvrdo“. Djeca, posebno dojenčad, po pravilu ne pene na ustima i ne grizu jezik. Napadi su rijetko praćeni mokrenjem, a san nakon napada je lakši, kratkotrajan i može ga potpuno izostati; nakon buđenja često nema pospanosti, usporenih reakcija ili letargije. Kod djece, češće nego kod odraslih, nakon epileptičnog napadaja, kada se svijest vrati, uočava se slabost u ruci i nozi s jedne strane (hemipareza). Postiktalnu hemiparezu karakteriše brz oporavak motoričke funkcije udova. Slabost u ruci i nozi traje nekoliko sati, a vrlo rijetko traje do jednog ili dva dana.
Jedan epileptički napad sam po sebi ne predstavlja opasnost po život djeteta. Međutim, ako se epileptički paroksizmi slijede jedan za drugim i ne prestaju sami, a njihovo trajanje doseže nekoliko sati i dana, ovo stanje se naziva epileptični status. Pojava statusa zahtijeva mjere reanimacije.
Pacijent iznenada gubi svijest. Nakon nekoliko sekundi vraća se svijest i osoba, kao da se ništa nije dogodilo, nastavlja radnju ili posao kojim je bila zauzeta prije napada. Međutim, neki pacijenti sa manjim napadima osjećaju i razumiju da im se nešto dogodilo. Male napadaje karakterizira visoka učestalost: od nekoliko puta do desetina i stotina puta dnevno.
Mali epileptički napadi su vrlo raznoliki, ali nema motoričke „bure“ ili padova, kao kod grand mal epileptičkog napada.
Kod djece prve godine života epileptički napadi se nazivaju infantilnim grčevima. Mogu imati karakter simptomatske (sekundarne) generalizirane epilepsije. Bolest se zasniva na različitim patološkim stanjima: poremećaji metabolizma aminokiselina, abnormalni razvoj moždane strukture - disgeneza, teška oštećenja mozga kao posljedica porođajna trauma, bivši intrauterini gladovanje kiseonikom(hipoksija) ili infektivne lezije mozak.
Afektivno-respiratorni paroksizmi
Kod mlađe djece i predškolskog uzrasta javljaju se takozvani mioklonični napadi: trenutni trzaji u rukama i licu, u kombinaciji s trenutnim nesvjesticama. Mioklonični paroksizmi mogu biti benigni, ali se javljaju i maligni simptomatski oblici.
Neke epileptičke napade karakterizira povezanost s ritmom sna i budnosti: ako se napadi javljaju prije ili neposredno nakon buđenja, govore o „epilepsiji buđenja“. Postoji tzv. noćna epilepsija“, kada se napadi javljaju samo tokom spavanja.
Neliječena epilepsija kod djece prvih godina života često dovodi do teških psihičkih poremećaja i razvoj govora. Međutim, mnogo ovisi o karakteristikama epileptičkog procesa. U nekim slučajevima se kod istog pacijenta može uočiti kombinacija nekoliko vrsta paroksizama, u ovom slučaju govore o polimorfnim napadima.
Afektivno-respiratorni paroksizmi kod djece zaslužuju pažnju, jer su ponekad predznaci epilepsije. Ovi napadi se razvijaju u pozadini intenzivnog plača, odnosno teške hiperventilacije, što izaziva razvoj epileptičkog napada. At mnogo plače disanje prestaje, nasolabijalni trokut postaje plav, dijete se često napinje ili, obrnuto, mlohavo, a ponekad se svijest može isključiti. Takvi napadi najčešće se javljaju kod djece prve godine života. Morate znati o njima kako biste na vrijeme kontaktirali neurologa i obavili neophodan pregled.
Genetske lezije N.s.
Mogu se naslijediti autosomno dominantno, autosomno recesivno i spolno vezano. Relativno izražena sistemičnost lezije N.s. u ovim oboljenjima nam omogućava da ih podijelimo u grupe sa dominantnim oštećenjem piramidalnog sistema, subkortikalnih formacija, malog mozga i njegovih veza te neuromišićnih bolesti. Najčešći su Daunova bolest, Shereshevsky-Turnerov sindrom, Klinefelterov sindrom itd. Nasljedna priroda niza kroničnih progresivnih degenerativnih bolesti nervnog sistema (na primjer, mijastenija gravis, siringomijelija) nije utvrđena.
Daunova bolest
Uzrokovana anomalijom hromozomskog skupa (promena broja ili strukture autosoma), čije su glavne manifestacije mentalna retardacija, osebujna izgled pacijenta i kongenitalne malformacije.
Osnova bolesti u velikoj većini slučajeva je trisomija na 21. paru hromozoma, odnosno umjesto dva postoje tri hromozoma, pa stoga sve ćelije sadrže 47 hromozoma [kariotip 47, XX (XY), + 21]. Budući da je učestalost rađanja djece sa D. b. naglo raste kod žena starijih od 35-40 godina, vjeruje se da dodatni 21. kromosom u većini slučajeva nastaje kao rezultat nedisjunkcije hromozoma tokom sazrijevanja ženske zametne stanice. U približno 1/3 slučajeva D. b. povezana s neraspadanjem hromozoma u muškoj reproduktivnoj ćeliji.
Karakterističan je spoljašnji izgled bolesnika: kose palpebralne pukotine, široki spljošteni most nosa, dodatni kožni nabor u unutrašnjem uglu oka (epikantus), poluotvorena usta, deformisana. uši, uvećan jezik sa hipertrofiranim papilama i dubokim žljebovima, visoko zasvođeno nepce; bjelkaste mrlje su često vidljive duž periferije šarenice; kratak vrat, stopala i ruke kratke i široke; prsti kao da su odsječeni, mali prst je skraćen i zakrivljen, ima jedan fleksijski nabor umjesto normalna dva; na dlanu se često nalazi poprečni nabor; na stopalima je povećan razmak između prvog i drugog prsta. Pacijenti su zakržljali od rođenja i počinju da drže podignutu glavu, sjede i hodaju do kasno. Zubi izbijaju kasno i pogrešnim redosledom. Seksualni razvoj naglo odloženo.
U kliničkoj slici bolesti dominiraju promjene na nervnom sistemu. Kod većine pacijenata smanjen je obim glave. Od prvih dana djetetovog života otkriva se mišićna hipotonija. Postoji strabizam, obično konvergirajući, slabost konvergencije, asimetrija inervacije lica, horizontalni nistagmus. Neka djeca sa D. b. otkrivaju se poremećaji koordinacije. Svi pacijenti imaju autonomno-endokrine poremećaje: suhu kožu, predispoziciju za gojaznost, dermatitis, crveni perzistentni dermografizam, degenerativne promjene na kostima. S godinama se javlja tendencija normalizacije mišićnog tonusa, poboljšava se i koordinacija pokreta.
Duševne poremećaje karakterizira uglavnom demencija tipa mentalne nerazvijenosti - oligofrenija, koja se otkriva već u prvoj godini života. Govor se kasno razvija leksikon loš, izgovor sa nedostacima. Karakterizira ih sporo razmišljanje, slaba sposobnost prebacivanja, pacijenti se lako gube u nepoznatom okruženju. Pažnja je nestabilna i lako se odvlači. Mehanička memorija je relativno dobro razvijena i imitacija je izražena. Emocije su slabo diferencirane, pacijenti su pasivni i zavisni, a karakterizira ih povećana sugestibilnost.
Zaključak
Posljednjih godina postalo je jasno da mnoge bolesti starije djece, pa čak i odraslih, imaju svoje porijeklo u djetinjstvu i često su kasna osveta za neprepoznatu i neliječenu patologiju novorođenčeta.
Mora se izvući jedan zaključak - biti pažljiv prema zdravlju bebe od trenutka začeća, blagovremeno otkloniti, ako je moguće, sve štetne posljedice po njegovo zdravlje, a još bolje, potpuno ih spriječiti. Ako se dogodi ovakva nesreća i otkrije se patologija nervnog sistema kod djeteta pri rođenju, potrebno je na vrijeme kontaktirati pedijatrijskog neurologa i učiniti sve da se beba potpuno oporavi.
Književnost
Bekhtereva N.P. Zdrav i bolestan ljudski mozak, M., 2000;
Bekhtereva N.P. i dr. Mehanizmi aktivnosti ljudskog mozga. Dio 1. -- Humana neurofiziologija, M., 2001;
Gusev E.I., Grechko V.E. i Burd G.S. Nervne bolesti, M., 2000;
Kostyuk P.G. Fiziologija centralnog nervnog sistema, Kijev, 2002;
Kuffler S. i Nichols J. Od neurona do mozga, trans. iz engleskog, M., 2001;
Nasljedne bolesti, ur. L.O. Badaljan, Taškent, 2004;
Objavljeno na Allbest.ru
Slični dokumenti
Sindrom kao skup simptoma ili karakteristične karakteristike. Oblici Downovog sindroma. Širenje patologije, uzroci njenog nastanka. Utjecaj starosti majke na vjerovatnoću Downovog sindroma kod djeteta. Pregled za otkrivanje smetnji u razvoju fetusa.
prezentacija, dodano 20.04.2012
Učestalost rađanja djece sa Downovim sindromom. Downov sindrom kao jedan od oblika oligofrenije uzrokovan abnormalnošću hromozomskog seta. Simptomi i klinička slika poremećaja. Istraživanje Downovog sindroma. Neuropsihološki podaci, poremećaji u radu analizatora.
prezentacija, dodano 18.05.2010
Klasični oblik hemoragijske bolesti novorođenčeta u akutnom periodu. Perinatalno oštećenje centralnog nervnog sistema hipoksičnog porekla, teško. Sindrom mišićne hipotonije i poremećaja kretanja, intrauterina infekcija.
anamneza, dodata 05.05.2014
Razvoj djece sa Downovim sindromom. Detekcija Downovog sindroma kod fetusa pomoću moćne ultrazvučne opreme. Simptomi Downovog sindroma, retardacija u psihomotorici i intelektualni razvoj. Preporuke za roditelje djece sa Downovim sindromom, rad sa njima.
prezentacija, dodano 24.04.2010
Istorija pojma "Downov sindrom". Uzroci bolesti, njeni oblici, spoljni znaci, njegov tretman i posljedice. Suština preimplantacijske genetske dijagnostike, dopler ultrazvuk, trodimenzionalna ultrazvuk i druge metode. Savjeti za trudnice.
prezentacija, dodano 22.03.2010
Konvulzivni sindrom kod djece je nespecifična reakcija djetetovog tijela na vanjske i unutrašnje podražaje, koju karakteriziraju iznenadni napadi mišićnih kontrakcija. Analiza uzroka konvulzivnog sindroma: disbalans elektrolita, endokrini poremećaji.
prezentacija, dodano 23.11.2015
Gonadni, ekstragonadalni i ekstrafetalni kongenitalne forme poremećaji seksualnog razvoja. Kromosomska patologija. Shereshevsky-Turnerov sindrom i “čista” ageneza gonada. Poremećaji segregacije X-hromozoma. Klinefelterov sindrom, nepotpuna maskulinizacija.
prezentacija, dodano 18.04.2016
Sindromi čiji je razvoj uzrokovan promjenama u broju ili strukturi kromosoma. Učestalost hromozomskih bolesti kod novorođenčadi. Downov sindrom, Patauov sindrom, Edwardsov sindrom. Anomalije kombinacije polnih hromozoma. Sindromi parcijalne monosomije.
prezentacija, dodano 01.06.2013
Kongenitalni razvojni poremećaj koji se manifestuje mentalna retardacija, poremećeni rast kostiju i druge fizičke abnormalnosti. Mogući razlozi Downov sindrom. Genetsko istraživanje. Po čemu se beba s Downovim sindromom razlikuje od druge djece?
sažetak, dodan 01.10.2009
Učestalost razvoja konvulzivnog sindroma. Perinatalna hipoksija, intrakranijalno krvarenje. Metabolički poremećaji praćeni hipoglikemijom. Genetski i urođene mane razvoj mozga. Klasifikacija napadaja kod novorođenčadi prema Volpeu.