Neuromišićne bolesti karakteriziraju disfunkcija voljnih mišića, gubitak ili smanjenje motoričke kontrole, što može nastati kao posljedica oštećenja samih mišića, ili biti sekundarne prirode - zbog disfunkcije neuromišićnog spoja, oštećenja perifernih nerava ili motornih neurona kičmena moždina. Klinička slika nekih neuromišićnih bolesti može sadržavati znakove oštećenja motoričkih jezgara moždanog stabla. Lezije u drugim područjima nervni sistem koje dovode do oštećenja motoričke kontrole, a posebno piramidalnog trakta, prema općeprihvaćenoj definiciji, ne spadaju u neuromišićne bolesti.
Najčešći simptomi neuromišićnih bolesti su slabost, smanjen volumen mišića (atrofija), nevoljni trzaji mišića, grčevi, utrnulost, trnci, itd. Oštećenje funkcije neuromišićnog spoja može uzrokovati spuštanje očnih kapaka (ptoza), dvostruki vid (diplopija) i drugi znakovi slabosti mišića koji se pogoršavaju kako dan odmiče. Neke bolesti mogu uticati na gutanje, pa čak i na disanje.
Bolesti mišića: simptomi
- Progresivne mišićne distrofije– genetski nasledna bolest mišića, čiji se simptomi obično javljaju u djetinjstvu ili djetinjstvo, rjeđe kod odraslih. Slabost mišića se postepeno povećava, posebno uočljiva u voljnim mišićima. U ovu grupu spadaju Beckerova mišićna distrofija, kongenitalna mišićna distrofija, distalna mišićna distrofija, Duchenneova mišićna distrofija (najčešći oblik mišićne distrofije kod djece), Emery-Dreyfusova mišićna distrofija, rameno-skapularna mišićna distrofija, miotonična mišićna distrofija (najčešća miotonična mišićna distrofija). mišićne distrofije kod odraslih), okulofaringealna miodistrofija.
- Inflamatorne miopatije– naziva se i miozitis, zasniva se na zapaljenom procesu koji dovodi do slabosti mišića, u njihovom nastanku je naglašena uloga autoimunih poremećaja, ponekad u kombinaciji s drugim autoimune bolesti. To uključuje dermatomiozitis, polimiozitis i miozitis inkluzijskog tijela.
- Mitohondrijalne miopatije– nastaju kao rezultat strukturnih ili biohemijskih defekata mitohondrija. Kearns-Sayreov sindrom, mioklonusna epilepsija sa „polomljenim crvenim vlaknima“, mitohondrijalna encefalomiopatija.
- Miotonija - kongenitalna miotonija ili Thomsenova bolest, distrofična miotonija, kongenitalna paramiotonija, neuromiotonija ili Isaacsova bolest
- Ostale miopatije – bolest centralnog jezgra, miotubudularna miopatija, nemalinska miopatija, periodična hiperkalemijska i hipokalemijska paraliza, endokrine miopatije
Bolesti neuromuskularnog spoja
Oni uzrokuju disfunkciju normalnog sinaptičkog prijenosa impulsa od nervnih završetaka do mišićnih vlakana. Bolest može biti zasnovana na autoimunom procesu.
- miastenija gravis
- Lambert-Eatonov sindrom
- kongenitalni miastenični sindrom
Bolesti perifernih nerava
- Mononeuropatije - oštećenje jednog živca, najčešći uzrok je kompresija ( tunelski sindromi), traumatske povrede
- Multiple mononeuropatije su multifokalne lezije perifernih nerava, obično povezane sa sistemskim ili infektivnim bolestima, paraneoplastičnim sindromima
- Polineuropatije su difuzne, simetrične lezije perifernih nerava, najčešće preovlađujuće u distalnim dijelovima; poremećaji osjetljivosti su gotovo uvijek prisutni u kliničkoj slici. Mogu biti akutne (Guillain-Barréov sindrom), kronične (kronična upalna demijelinizirajuća polineuropatija), stečene (toksične, dijabetičke, paraneoplastične) ili nasljedne (peronealna mišićna atrofija ili Charcot-Marie-Toothova bolest, Dejerine-Sottasova bolest, Friedrexia).
- Pleksopatije su lezije pleksusa gornjih i donjih ekstremiteta (brahijalni i lumbosakralni), čiji su najčešći uzrok traumatski ili kompresivni efekti
- Radikulopatije i poliradikulopatije - lezije motoričkih ili senzornih spinalnih korijena
Bolesti motorni neuron
Progresivni degenerativni poremećaj motornih neurona koji najviše rezultira poremećenom motoričkom kontrolom gornjih ili donjih ekstremiteta, kao i bulbarnim poremećajima. Najčešće počinju u srednjim godinama, simptomi mogu uključivati slabost u udovima, otežano gutanje, govor, hod, slabost mišića lica i grčeve mišića. Ova grupa uključuje, posebno:
- strana amiotrofična skleroza(BASS)
- spinalna mišićna atrofija odraslih
- spinalna mišićna atrofija kod novorođenčadi ili Werdnig-Hoffmannova bolest
- juvenilna spinalna mišićna atrofija ili Kugelberg-Welanderova bolest
- bulbospinalna mišićna atrofija ili Kennedyjeva bolest
Dijagnoza dijagnosticira se na osnovu anamneze bolesti, detaljnim neurološkim pregledom, u većini slučajeva koristi se elektromiografska (EMG) studija, u kombinaciji sa oštećenjem centralnog motornog neurona ili za njegovo isključenje može se koristiti transkranijalna magnetna stimulacija, ako su nasljedni oblici se sumnja, radi se DNK analiza, autoimuna priroda procesa zahtijeva određivanje specifičnih antitijela, može se uraditi biopsija mišićnog područja, u slučaju primarnih mišićnih lezija provjerava se nivo kreatin fosfokinaze (CPK), u U poslednje vreme takođe dobija na popularnosti ultrazvučni pregled mišića i perifernih nerava. Dijagnostički algoritam, izbor dodatna istraživanja zavise od karakteristika kliničkog obrasca i lokalizacije lezije - mišića, živaca, pleksusa, korijena, motornih neurona.
NASLJEDNE BOLESTI NERVNOG SISTEMA
PREDAVANJE 16
Degenerativne bolesti koje prvenstveno pogađaju neuromišićni sistem čine najveću grupu svih nasljednih bolesti.
Rezultati elektrofizioloških i biohemijskih studija izuzetno su važni i često odlučujući u dijagnostici neuromišićnih bolesti. Jednako je velika važnost patomorfoloških nalaza. Studija biopsije mišića u svetlosni mikroskop pomaže u razlikovanju miogene atrofije od neurogene atrofije. Histohemijski pregled je neophodan za identifikaciju metaboličke lezije mišića, a elektronska mikroskopija je otkrila čitavu veliku klasu bolesti - neprogresivne miopatije.
Progresivna mišićna distrofija. Pojam mišićna distrofija odnosi se na grupu genetski uvjetovanih poremećaja koje karakteriziraju progresivne degenerativne promjene u mišićnim vlaknima bez primarne patologije perifernog (donjeg) motornog neurona.
Razni oblici razlikuju se jedni od drugih po vrstama nasljeđivanja, vremenu početka procesa, prirodi i brzini njegovog tijeka, jedinstvenoj topografiji mišićne atrofije, prisutnosti ili odsutnosti pseudohipertrofije i povlačenja tetiva i drugim karakteristikama.
Većina mišićnih distrofija je dobro klinički proučavana i Detaljan opis napravljena krajem prošlog veka. No, uprkos gotovo stoljetnoj povijesti proučavanja mišićnih distrofija, pitanja njihove patogeneze i liječenja ostaju neriješena do danas. Velike nade polažu se u molekularnu genetiku, uz pomoć koje je utvrđena lokacija gena za mnoge nozološke oblike.
Dijagnoza mišićne distrofije često je vrlo teška. Postoji velika varijabilnost kliničkih manifestacija, a mali broj članova porodice otežava određivanje načina nasljeđivanja.
Karakterističan motorički defekt kod pacijenata sa mišićnom distrofijom je „pačji” hod: pacijent hoda galajući s jedne strane na drugu. Povezan je uglavnom sa slabošću glutealnih mišića, prvenstveno srednjih i malih mišića, koji fiksiraju karlicu u odnosu na femur. Kao rezultat, bolest uzrokuje naginjanje karlice prema nozi koja ne podupire (Trendelenburgov fenomen) i kompenzacijski nagib trupa u suprotnom smjeru (Duchenneov fenomen). Prilikom hodanja strana nagiba se stalno mijenja. Ove promjene se mogu provjeriti i Trendelenburgovim testom tako što se od pacijenta traži da podigne jednu nogu, savijajući je pod pravim kutom u zglobovima koljena i kuka: karlica sa strane podignute noge se spušta (i ne podiže se kao normalno) zbog slabosti gluteus medius mišića potporne noge.
Rising from horizontalni položaj, bolesnik sa izraženom slabošću mišića proksimalnih mišića teško se prevrće na trbuh, zatim, oslonjen rukama na pod, ustaje na sve četiri, a zatim se, oslanjajući se rukama na potkoljenice, zatim na butine, postepeno ispravlja. Ovaj fenomen "svoje dobiti" naziva se Goversov manevar. Često se povezuje sa slabošću mišića gluteusa maximusa.
Duchenneova mišićna distrofija. Duchenneova pseudohipertrofična mišićna distrofija javlja se češće od svih ostalih bolesti mišićnog sistema (30 na 100.000 živorođene djece). Karakterizira ga rani početak i maligni tok. Klasična slika se manifestuje promjenom hoda djeteta u dobi od 2-5 godina; u dobi od 8-10 godina djeca već otežano hodaju; do 14-15 godina su kao pravilo, potpuno imobiliziran. Djeca imaju više rane godine početni simptomi se manifestiraju zakašnjelim motoričkim razvojem: kasnije počinju hodati i ne mogu trčati ili skakati. Bolesnici umiru u 2-3. deceniji života.
Jedan od prvih znakova bolesti je zadebljanje mišića lista i postupno povećanje njihovog volumena zbog pseudohipertrofije. Atrofiju mišića bedara i karličnog pojasa često maskira dobro razvijeno potkožno masno tkivo. Postupno, proces ide prema gore i širi se izvan ramenog pojasa, leđnih mišića, a zatim i do proksimalnih dijelova ruku.
IN terminalni stepen mišićna slabost može se proširiti na mišiće lica, ždrijela i respiratorne mišiće.
U uznapredovaloj fazi bolesti postoje takvi karakteristični simptomi, kao “pačja šetnja”; naglašena lumbalna lordoza, pterygoid blades, simptom “labavih ramenih pojaseva”. Tipične su rane mišićne kontrakture i retrakcije tetiva, posebno Ahilove tetive. Rano nestaju refleksi koljena, a potom i refleksi iz gornjih ekstremiteta.
Pseudohipertrofija se može razviti ne samo u mišićima lista, već iu glutealnim, deltoidnim, trbušnim mišićima i mišićima jezika. Vrlo često srčani mišić pati od neke vrste kardiomiopatije. Otkrivaju se poremećaji u ritmu srčane aktivnosti, proširenje granica srca, tupost tonova i promjene EKG-a. Najviše je akutnog zatajenja srca uobičajen razlog smrtni ishodi kod Duchenneove mišićne distrofije. Na obdukciji se nalazi fibroza i masna infiltracija srčanog mišića.
Često se uočavaju poremećaji gastrointestinalnog motiliteta.
Uobičajeni simptom je smanjenje inteligencije. Zanimljivo je da je u nekim porodicama mentalna retardacija izražena, u drugim relativno umjerena. Promjena višeg mentalne funkcije obično ne napreduje i nije u korelaciji sa ozbiljnošću mišićnog defekta. To se ne može objasniti samo pedagoškim zanemarivanjem bolesne djece koja su rano isključena iz dječjih grupa i ne pohađaju ih. vrtić i školu zbog motoričkih mana. CT i MRI često otkrivaju cerebralnu atrofiju, vjerojatno povezanu s oštećenjem prenatalnog razvoja mozga.
Djeca često razvijaju adiposogenitalni sindrom, a ponekad i druge znakove endokrine insuficijencije. Često se javljaju promene na koštanom sistemu: deformacije stopala, prsa, kralježnica, difuzna osteoporoza.
Prepoznatljiva karakteristika Duchenneov oblik je visok stepen hiperenzimija je već na ranim fazama razvoj procesa. Dakle, nivo enzima specifičnog za mišićno tkivo - kreatinin fosfokinaze - u krvnom serumu može premašiti desetine, pa čak i stotine puta normalni indikatori. Naglo (10-100 puta) povećanje kreatinin fosfokinaze (CPK) u neuromuskularnoj patologiji trebalo bi prije svega potaknuti raspravu sledeće bolesti: Duchenneova bolest, Beckerova bolest, poliomiozitis i dermatomiozitis, paroksizmalna mioglobulinurija, distalna miodistrofija. Samo u uznapredovalim stadijumima bolesti stepen hiperenzimemije se postepeno smanjuje. Postoje izvještaji o povećanju CK u fazi intrauterinog razvoja.
Duchenneova mišićna distrofija se prenosi na X-vezan recesivan način. Gen je lokaliziran na kratkom kraku X hromozoma. Učestalost mutacije gena je prilično visoka (30%), što objašnjava veliki broj sporadični slučajevi.
Mutacija (najčešće delecija) dovodi do seksualnog ili gotovo potpunog odsustva genskog proizvoda - strukturnog proteina distrofije. Fiziološka uloga distrofije nije u potpunosti utvrđena. Nalazi se u visokim koncentracijama u regiji sarkoleme, očito igrajući određenu ulogu u održavanju integriteta ove membrane. Odsustvo distrofije uzrokuje strukturne promjene u sarkolemi, što zauzvrat dovodi do gubitka intracelularnih komponenti i povećanog priliva kalcija, što u konačnici dovodi do odumiranja miofibrila. Vjeruje se da je nedostatak distrofije u sinaptičkim zonama kortikalnih neurona uzrok mentalne retardacije.
Za medicinsko genetičko savjetovanje veoma je važno utvrditi heterozigotnu kolekciju. Kod Duchenneove mišićne distrofije kod heterozigota u približno 70% slučajeva otkrivaju se subklinički i ponekad očigledni znakovi mišićne patologije - određeno zadebljanje, pa čak i povećanje mišića lista, brzi zamor mišića tokom fizičke aktivnosti, promjene EMG-a i patomorfološkog pregleda. biopsije mišića. Najčešće, heterozigotni nosioci pokazuju povećanje aktivnosti kreatinin fosfokinaze.
Ako postoji klinička slika Duchenneove mišićne distrofije kod žena, prvo treba isključiti mogućnost abnormalnosti na X hromozomu - Shereshevsky-Turnerov sindrom (TS), Morrisov sindrom (XY) ili mozaicizam za ove sindrome.
Duchenneova mišićna distrofija, koja počinje da se razvija u prenatalnom periodu, u suštini je kongenitalna miopatija i može se dijagnosticirati ubrzo nakon rođenja izvođenjem mišićne biopsije i određivanjem aktivnosti CPK.
Beckerova mišićna distrofija. Zajedno sa teškim maligni oblik Postoji benigni oblik X-vezane Duchenneove mišićne distrofije - Beckerova bolest. Po kliničkim simptomima, vrlo je sličan Duchenneovom obliku, ali u pravilu počinje kasnije - sa 10-15 godina, teče blago, pacijenti ostaju radno sposobni dugo vremena, u dobi od 20 godina. 30 godina i kasnije još uvijek mogu hodati. Plodnost se ne smanjuje, pa se bolest ponekad može pratiti u nekoliko generacija porodice: bolestan čovjek preko kćerke prenosi bolest na unuka („djedov efekat“). Početni simptomi, kao i kod Duchenneove bolesti, manifestiraju se kao slabost u mišićima karličnog pojasa, zatim u proksimalnim dijelovima donjih ekstremiteta. Hod pacijenata se mijenja, imaju poteškoća pri penjanju uz stepenice ili ustajanju sa niskog sjedišta. Karakteristična je pseudohipertrofija mišića lista. Retrakcija petne (Ahilove) tetive je manje izražena nego kod Duchenneove bolesti.
Kod ovog oblika nema intelektualnih smetnji, kardiomiopatija je odsutna ili je blago izražena.
Kao i kod drugih X-vezanih miodistrofija, u Beckerovom obliku aktivnost CPK značajno raste, iako u manjoj mjeri nego kod Duchenneove bolesti, ne prelazi 5000 jedinica. Gen za Beckerovu bolest, poput Duchenneove bolesti, lokaliziran je u kratkom kraku X hromozoma; vjerovatno je da su oba lokusa blisko povezana ili alelna. Za razliku od Duchenneove bolesti, kod koje praktički nema distrofije, kod Beckerove bolesti se sintetizira abnormalna distrofija. Razlike se također nalaze u biopsiji mišića. Kod Beckerove mišićne distrofije mišićna vlakna su obično nezaobljena, a hijalinska vlakna, karakteristična za Duchenneovu mišićnu distrofiju, uočavaju se izuzetno rijetko.
Landouzy-Dejerine miodistrofija (facioskapulohumeralna miodistrofija). Bolest se prenosi na autosomno dominantan način sa visokom penetracijom, ali donekle varijabilnom ekspresivnošću. Mnogo je rjeđa od Duchenneove mišićne distrofije (0,4 na 100 hiljada stanovnika). Vjeruje se da je gen za ovu bolest lokaliziran na 4. kromosomu. Žene obolijevaju češće od muškaraca (3:1), Fizičko preopterećenje, intenzivni sportovi, kao i neracionalni fizioterapija može doprinijeti težem toku bolesti.
Landouzy-Dejerine mišićna distrofija - relativno povoljna trenutni oblik patologija mišića. Počinje u dobi od oko 20 godina, ponekad i kasnije. Međutim, u porodične slučajeve bolesti, kada se može pratiti dinamika mlađih članova porodice, moguće je uočiti neku slabost mišića, na primjer mišića lica, u ranijoj dobi.
Mišićna slabost i atrofija prvo se javlja u mišićima lica ili ramenog pojasa. Postepeno se ovi poremećaji šire na mišiće proksimalnih ruku, a zatim i na mišiće donjih udova. U većini slučajeva prvo su zahvaćeni mišići prednje površine nogu (sa razvojem pada stopala), zatim mišići proksimalnih nogu. U jeku bolesti jako su zahvaćeni orbitalni mišići oka i usta, pectoralis major, serratus anterior i donji dijelovi mišića trapeza. latissimus mišića leđa, biceps, triceps brachii mišići. Karakteristično izgled pacijenti: tipično lice miopatija sa "poprečnim osmehom" ("Gioconda osmeh"), protruzija gornja usna(„tapirske usne“), izražene lopatice u obliku krila, osebujna deformacija grudnog koša sa spljoštavanjem u anteroposteriornom smjeru i rotacijom prema unutra rameni zglobovi. Često postoji asimetrija lezije, čak i unutar istog mišića (npr. orbicularis mišić usta). Može se uočiti pseudohipertrofija listova, deltoidnih mišića, a ponekad i mišića lica. Kontrakcije i retrakcije su umjereno izražene. Tetivni refleksi dugo vrijeme su očuvane, ali se ponekad smanjuju već u ranoj fazi.
Znakovi oštećenja srčanog mišića rijetko se otkrivaju. Aktivnost enzima u serumu je blago povećana i može biti normalna. Inteligencija ne trpi. Očekivano trajanje života u većini slučajeva se ne smanjuje. Zanimljivo je da EMG kod Landouzy-Dejerine miodistrofije često nije sasvim tipičan za mišićni nivo lezije. Kod nekih pacijenata (članova iste porodice) može doći do smanjenja amplitude biopotencijala, krivulje interferencije; kod drugih, naprotiv, smanjenje frekvencije i hipersinhrone aktivnosti, ponekad sa tipičnim palisadnim ritmom. Treba biti svjestan spinalne varijante koja oponaša Landouzy-Dejerine bolest.
Erb-Roth mišićna distrofija (mišićna distrofija udova-pojasa). Prenosi se autosomno recesivno i podjednako su zahvaćena oba pola. Početak bolesti u većini slučajeva datira od sredine 2. decenije života (14-16 godina), ali se opisuje kao rani, pseudo-Duchenneov oblik, kada se prvi simptomi javljaju prije 10. godine života. a bolest je teška i kasna verzija sa početkom nakon 30 godina.
Tijek bolesti može biti brz ili sporiji, u prosjeku potpuni invaliditet nastupa 15-20 godina od pojave prvih simptoma. Miodistrofija počinje ili oštećenjem mišića karličnog pojasa i proksimalnih nogu (Leiden-Moebiusov oblik), ili od ramenog pojasa (Erb oblik). U nekim slučajevima, rameni i karlični pojas su zahvaćeni istovremeno. Mišići leđa i abdomena prilično pate. Pacijenti imaju karakterističan "pačji" hod, otežano ustajanje iz ležećeg ili sedećeg položaja, naglašeno lumbalna lordoza. U većini slučajeva mišići lica nisu zahvaćeni. Kontrakture i pseudohipertrofije su neuobičajene za ovaj oblik. Može doći do atrofije terminala i retrakcije tetive. Inteligencija je obično očuvana. Srčani mišić uglavnom nije zahvaćen. Nivoi enzima u serumu su obično povišeni, ali ne tako dramatično kao kod X-vezane mišićne distrofije. Postoje indicije da muški pacijenti imaju viši nivo CPK od ženskih pacijenata. Postoji značajna razlika u ekspresivnosti mutantnog gena u različitih članova porodica - zajedno sa teškim kliničku sliku Mogu postojati relativno blagi, pa čak i zamagljeni klinički simptomi. Smrt obično nastupa od plućnih komplikacija.
Budući da se posebno lako oponaša klinika miodistrofije pojasa udova neuromuskularne bolesti drugačije prirode, potrebno je, posebno u sporadičnim slučajevima i kasnom nastanku bolesti, izvršiti detaljan klinički pregled kako bi se isključila spinalna amiotrofija, polimiozitis, metaboličke, endokrine, toksične, lijekovima inducirane, karcinomatozne miopatije. U prošlosti je postojala jasna prevelika dijagnoza ovog oblika mišićne distrofije.
Liječenje mišićnih distrofija. Terapijske mogućnosti za mišićne distrofije su vrlo ograničene. Etiološki i patogenetski tretman praktično nepostojeće. Simptomatsko liječenje prvenstveno je usmjereno na sprječavanje razvoja kontraktura, održavanje postojeće mišićne snage i, eventualno, blago smanjenje stope razvoja atrofije. Glavni zadatak je maksimizirati period tokom kojeg se pacijent može samostalno kretati, jer ležeći položaj kontrakture, skolioza i respiratorni poremećaji se brzo povećavaju. Medicinski kompleks treba uključivati terapeutske vježbe, masažu, ortopedske mjere i terapiju lijekovima.
Terapeutska gimnastika se sastoji od pasivnih i aktivnih pokreta koji se izvode u svim zglobovima u različitim položajima: stojeći, sedeći, ležeći, sa različitim položajima udova. Poželjno je izvoditi aktivne pokrete u izometrijskom načinu. Časove gimnastike treba izvoditi redovno nekoliko puta dnevno. U isto vrijeme, treba biti oprezan protiv prekomjernog vježbanja, posebno kada je praćeno prenaprezanjem mišića. Bitan(posebno nakon imobilizacije pacijenta) imati vježbe disanja.
Ortopedske mjere konzervativne (specijalne udlage) i operativne prirode (ahilotomija, presek potkoljenični mišić), usmjerene na ispravljanje kontraktura i nastalih patoloških stavova udova, također imaju za cilj očuvanje sposobnosti samostalnog kretanja. U svakom slučaju potrebno je individualno odmjeriti očekivane koristi i moguća šteta od operacije. Treba uzeti u obzir da često (posebno kod teške hiperlordoze i slabosti mišića kvadricepsa femorisa) ekvinovarusni položaj stopala ima kompenzatornu vrijednost i nakon, na primjer, ahilotomije, pacijent može biti potpuno imobiliziran. Za razvoj kontraktura preporučuje se lagano istezanje mišića do 20-30 puta dnevno, nakon čega slijedi stavljanje udlage tokom spavanja.
Terapija lekovima uključuje propisivanje metaboličkih lijekova koji imaju za cilj nadoknaditi nedostatak energije i proteina, ali je njihova efikasnost vrlo sumnjiva. Koriste se antagonisti kalcija (zbog defekta stanične membrane identificiranog kod Duchenneove bolesti, što dovodi do povećanog ulaska kalcija u ćeliju), imunomodulatori, spojevi koji sadrže fosfor (ATP, fosfaden), vitamin E (100 mg oralno 3 puta dnevno). dan). Pokazalo se da kod Duchenneove bolesti upotreba prednizolona (0,75 mg/kg dnevno) može dramatično povećati snagu mišića, ali taj učinak ne traje duže od godinu dana i općenito ne utječe na ishod bolesti. Zbog ozbiljnog nuspojave, nastale laži dugotrajna upotreba lijeka, njegova upotreba je neprikladna. Procjene učinka anaboličkih steroida su kontroverzne i često se povezuje s njihovom upotrebom neopravdani rizik. Prilikom procjene djelovanja određenih lijekova na Duchenneovu bolest, treba uzeti u obzir da uz umjerenu težinu bolesti kod pacijenata u dobi od 3-6 godina može doći do relativne stabilizacije stanja povezane s razvoj uzrasta mišićnog sistema, sticanje motoričkih vještina, koje u određenoj mjeri mogu privremeno nadoknaditi kontinuirani degenerativni proces.
Definisana vrijednost ima korekciju ishrane pacijenta, preporučuje se dijeta sa visokim sadržajem proteina i niskim sadržajem masti i niskim sadržajem kalorija sa optimalnim sadržajem vitamina i mikroelemenata. Psihološka podrška pacijentu, kontinuirana edukacija i pravilno stručno vođenje igraju važnu ulogu.
Neuromuskularne bolesti (NMD) su najveća grupa nasljednih bolesti, koje se zasnivaju na genetski uvjetovanim oštećenjima prednjih rogova kičmene moždine, perifernih živaca i skeletnih mišića.
Neuromuskularne bolesti uključuju:
1) progresivne mišićne distrofije (primarne miopatije);
2) spinalne i neuralne amiotrofije (sekundarne miopatije);
3) kongenitalne neprogresivne miopatije;
4) neuromuskularne bolesti sa miotoničkim sindromom;
5) paroksizmalna mioplegija;
6) mijastenija gravis.
15.2. Progresivne mišićne distrofije (primarne miopatije)
Progresivna mišićna distrofija (PMD), ili primarne miopatije, karakteriziraju degenerativne promjene u mišićnom tkivu.
Patomorfološke promjene kod PMD-a karakterizira ih stanjivanje mišića, njihova zamjena masnim i vezivnim tkivom. U sarkoplazmi se otkrivaju žarišta žarišne nekroze, jezgra mišićnih vlakana su raspoređena u lance, a mišićna vlakna gube poprečne pruge.
Pitanja patogeneze ostaju neriješena do danas. Miopatija se zasniva na defektu membrane mišićne ćelije. Velike nade polažu se u molekularnu genetiku.
Različiti oblici miopatije razlikuju se po vrsti nasljeđivanja, vremenu početka procesa, prirodi i brzini njegovog tijeka i topografiji mišićne atrofije.
Miopatije se klinički karakteriziraju slabošću mišića i atrofijom. Postoje različiti oblici PMD-a.
15.2.1. Duchenneova mišićna distrofija (pseudohipertrofični oblik PMD-a)
Najčešći je od svih PMD (30:100.000). Ovaj oblik karakterizira rani početak (2-5 godina) i maligni tok, koji pogađa uglavnom dječake. Duchenneova miopatija se nasljeđuje na X-vezani recesivni način. Patološki gen je lokaliziran u kratkom kraku hromozoma (X ili hromozom 21).
Mutacija gena je prilično visoka, što objašnjava značajnu učestalost sporadičnih slučajeva. Mutacija (najčešće delecija) gena dovodi do izostanka distrofina u membrani mišićne ćelije, što dovodi do strukturnih promjena u sarkolemi. Ovo potiče oslobađanje kalcija i dovodi do smrti miofibrila.
Jedan od prvih znakova bolesti je otvrdnuće mišića lista i postupno povećanje njihovog volumena zbog pseudohipertrofije. Proces je odozdo prema gore. Uznapredovali stadijum bolesti karakteriše „pačji“ hod, pacijent hoda, gegajući se s jedne na drugu stranu, što je uglavnom zbog slabosti glutealnih mišića.
Kao rezultat, dolazi do nagiba zdjelice prema nozi koja ne podupire (Trendelenburgov fenomen) i kompenzacijskog nagiba trupa u suprotnom smjeru (Duchenneov fenomen). Prilikom hodanja strana nagiba se stalno mijenja. Ovo se može testirati u Trendelenburgovom položaju tražeći od pacijenta da podigne jednu nogu, savijajući je pod pravim kutom u zglobu koljena i kuka: karlica na strani podignute noge je spuštena (a ne podignuta kao normalno) zbog slabost gluteus medius mišića potporne noge.
Kod Duchenneove miopatije često se opaža izražena lordoza, krilate lopatice, tipične mišićne kontrakture i rani gubitak refleksa koljena. Često je moguće otkriti promjene na koštanom sistemu (deformacije stopala, grudnog koša, kičme, difuzna osteoporoza). Može doći do smanjenja inteligencije i raznih endokrinih poremećaja (adiposogenitalni sindrom, Itsenko-Cushingov sindrom). Do dobi od 14-15 godina pacijenti su obično potpuno imobilizirani; u terminalnoj fazi slabost se može proširiti na mišiće lica, ždrijela i dijafragme. Najčešće umiru u 3. deceniji života od kardiomiopatije ili dodatka interkurentnih infekcija.
Posebnost Duchenneove miopatije je naglo povećanje specifičnog mišićnog enzima - kreatin fosfokinaze (CPK) za desetine i stotine puta, kao i povećanje mioglobina za 6-8 puta.
Za medicinsko genetičko savjetovanje važno je uspostaviti heterozigotnu nosivost. Kod 70% heterozigota utvrđuju se subklinički i klinički znaci mišićne patologije: zadebljanje i povećanje mišića lista, brzi zamor mišića tokom fizičke aktivnosti, promjene u biopsijama mišića i biopotencijala prema EMG podacima.
Strana 44 od 44
Uključeni su skeletni mišići patološki proces za razne degenerativne, metaboličke i inflamatorne bolesti. U većini slučajeva dolazi do degeneracije mišićna vlakna, a kod kroničnih oblika - njihova zamjena vezivnim tkivom i masnoćom. Proksimalne mišićne grupe su značajnije oštećene od distalnih, kao i donji ekstremiteti u odnosu na gornje. Bolesno dijete ima takozvani pačji hod i ne može trčati, penjati se uz stepenice ili ustati ako je u sjedećem položaju. Njegovi tetivni refleksi su depresivni, stepen njihovog izumiranja je proporcionalan stepenu slabljenja mišićne snage. Osetljivost nije narušena.
Dijagnostički vrijedne laboratorijske metode uključuju određivanje aktivnosti enzima, posebno kreatin fosfokinaze, u serumu. Ovaj enzim, koji katalizuje reakciju: fosfokreatin + ADP-kreatin + ATP, prisutan je uglavnom u moždanim ćelijama i mišićnom tkivu. Kod nekih difuznih mišićnih bolesti, posebno kod mišićne distrofije, njegove suvišne količine prodiru u međućelijski prostor i krv. Pacijenti obično imaju povećanu aktivnost serumske laktat dehidrogenaze i glutamičke oksaloacetatne transaminaze, ali njihova široka distribucija u drugim tkivima, uključujući jetru, smanjuje specifičnost testa. Obično je potrebna biopsija mišićnog tkiva da bi se razjasnila dijagnoza.
Upalne bolesti mišića. Upala mišićnog tkiva prati neke infekcije, posebno trihinelozu, toksoplazmozu i one uzrokovane Coxsackie virusom. Često je komponenta kolagenskih bolesti, uključujući dermatomiozitis, eritematozni lupus, periarteritis nodosa i reumatoidni artritis.
Polimiozitis. Difuzna izolirana upala mišića nepoznate etiologije naziva se polimiozitis. Karakterizira ga brzi progresivni tok, slabost i bol u proksimalnim mišićnim grupama. Često su u proces uključeni mišići vrata, što otežava djetetu da podigne glavu i zadrži je u tom položaju. Laboratorijski znakovi upale mišića uključuju povećanje ESR-a i broja bijelih krvnih stanica. Međutim, njihov nedostatak ne isključuje polimiozitis. Nivoi enzima u serumu su obično povišeni. Biopsijom mišića utvrđuje se degeneracija i djelomična regeneracija vlakana i njihova infiltracija limfoidnim stanicama. Teško je razlikovati polimiozitis od mišićne distrofije i dermatomiozitisa. Može predstavljati atipična forma dermatomiozitis, iako je histološka slika za ova dva stanja nešto drugačija: dermatomiozitis karakterizira vaskulitis, koji obično nema kod polimiozitisa. Prognoza za potonje je nešto povoljnija. Liječenje kortikosteroidima je praćeno efektom, ali kada se oni prekinu, može doći do recidiva.
Progresivni miozitis ossificans. Etiologija ove rijetke bolesti vezivnog tkiva i mišića nije poznata. Izvještava se da pogađa braću i sestre, uključujući blizance, i prenosi se na krvne srodnike u pravoj liniji. Pretpostavlja se da se bolest nasljeđuje autosomno dominantno. Dječaci obolijevaju 2-3 puta češće od djevojčica.
Patološki znaci zavisi od stadijuma bolesti. U ranim fazama, lokalna oteklina i infiltrati upalnih stanica nalaze se u mišićima i tetivama. Kasnije se područja upale zamjenjuju granulacijskim tkivom i na kraju se u lezijama formiraju područja hrskavice i koštanog tkiva.
Gotovo 75% bolesne djece ima dijagnozu urođene mane razvoj, najčešće nerazvijenost prstiju i ankiloza falangi prvih prstiju i nerazvijenost prvih prstiju, polidaktilija, zakrivljenost prstiju, sindaktilija (noge), deformacija uši, gluvoća, nedostatak zuba. Isti urođeni defekti mogu biti prisutni i kod srodnika pacijenta kod kojih nije razvijena progresivna bolest vezivnog tkiva i mišića. Starost u kojoj može početi osifikativna miozitis varira od rođenja do starijeg djetinjstva. Obično se razlikuju u tri stadijuma: 1) na mestima lakših lokalnih povreda pojavljuju se ograničeni, često topli i mekani na dodir testasti otoki mekih tkiva; 2) nakon nekoliko dana simptomi upale nestaju, a lezija se stvrdne; 3) dolazi do okoštavanja zahvaćenog područja. Nove lezije se pojavljuju periodično, uglavnom u predelima vrata i leđa. Primarni simptom Tortikolis se može razviti ako se proces razvio u sternokleidomastoidnom mišiću. Osifikacija se na kraju širi na mnoge tetive i ligamente. Javlja se ankiloza kičme i zglobova ruku i nogu (sl. 21-5). Upala se može proširiti na temporomandibularne zglobove, otežavajući pokrete žvakanja. Koštane mamuze mogu viriti kroz kožu. U adolescenciji, bolest često dovodi do potpune nepokretnosti i smrti zbog respiratorna insuficijencija i prestanak disanja, iako postoje izvještaji o preživljavanju. Kod myositis ossificans postoji visok rizik od razvoja osteogenog sarkoma.
Rice. 21-5. Dijete s progresivnim miozitisom ossificans (tipično držanje s ukočenošću mišića vrata i leđa).
Ponekad je patološki proces ograničen na mjesto prethodne ozljede mekog tkiva (miositis ossificans circumscripta). Široko rasprostranjena kalcifikacija mišićnog tkiva može se pojaviti i kod kroničnog polimiozitisa i dermatomiozitisa.
rezultate laboratorijske metode studije nemaju dijagnostičku vrijednost.
Nivoi kalcijuma, fosfora, alkalne fosfataze i aktivnosti kreatin fosfokinaze i drugih enzima u serumu ostaju normalni. Koštano tkivo na mjestu ozljede ne razlikuje se po strukturi od norme.
Postojeće metode tretmani su nezadovoljavajući. U nekim slučajevima primećeno je usporavanje progresije bolesti primenom ACTH i drugih kortikosteroida. Njihova uloga u krajnji rezultat tretman je upitan.
Endokrine i metaboličke miopatije. Miopatija kod hipertireoze je prilično rijetka komplikacija. Karakterizira ga ptoza i bilateralna pareza. mišiće lica i mišiće proksimalnih udova. U ovom slučaju, neki simptomi hipertireoze mogu biti maskirani slabošću mišića, ali tahikardijom, pojačanim znojenjem i pojačanim štitne žlijezde. Refleksi tetiva, za razliku od mnogih drugih oblika miopatije, ostaju normalni. Nakon korekcije hipertireoze, slabost mišića postepeno nestaje.
Miopatija kod hipotireoze. Hipotireoza kod dojenčadi može biti povezana sa slabošću mišića i hipotenzijom. Kod starije djece s miksedemom kontrakcije i relaksacije mišića se usporavaju, au nekim slučajevima se opaža mišićna hipertrofija (Debreu-Semelen sindrom). Kombinacija znakova kao što su slabost i hipertrofija mišića ukazuje na mišićnu distrofiju.
Miopatija tokom terapije kortikosteroidima. Može zakomplikovati Itsenko-Cushingovu bolest, ali se češće razvija kada se liječi velikim dozama sintetičkih steroida. Posebno je uočljiva slabost u mišićima karličnog pojasa, koja se manifestuje geganjem (pačjim) hodom, otežanim penjanjem uz stepenice i pokušajem ustajanja iz sjedećeg položaja. Nema refleksa koljena. Može doći do stanjivanja mišića. Miopatske promjene u mišićnom tkivu obično su beznačajne čak i uz jaku slabost. Mišićna snaga se polako oporavlja nakon povlačenja kortikosteroida (tokom nekoliko mjeseci).
Miopatija kod hiperparatireoze. Hiperparatireoza može biti povezana sa slabošću i hiporefleksijom uzrokovanom hiperkalemijom. Obično nestaju brzo nakon paratiroidektomije.
Nedostatak karnitina (lipidna miopatija) prati nakupljanje velikih količina lipida u mišićima i, kao rezultat, poremećaj u opskrbi energijom potonjih. Karnitin je jedna od obaveznih komponenti sistema koji osigurava transfer masne kiseline sa dugim lancem od citosola do mitohondrija, gde prolaze kroz oksidaciju. Slabost mišića se razvija u dva oblika nedostatka karnitina.
Nedostatak karnitina u mišićima klinički je predstavljen progresivnom slabošću njihovih proksimalnih grupa, češće kod školaraca i adolescenata. Ponekad je slabost povremeno i u kombinaciji sa mioglobinurijom. U teškim slučajevima može doći do paralize respiratornih mišića. Nivoi enzima u serumu (kreatin kinaze i aldolaze) se povećavaju. Elektromiogram otkriva nespecifične promjene karakteristične za miopatiju. U biopsiji mišića možete vidjeti veliki broj masnih kapljica. Nivoi karnitina u serumu se ne mijenjaju, ali se nivoi karnitina u mišićima smanjuju. Prepoznavanje patologije je neophodno jer se može izliječiti. Često se pogrešno smatra mišićnom distrofijom. Efekat se može javiti nakon oralne primjene 100 mg/(kg/dan) karnitina. U nekim slučajevima, liječenje kortikosteroidima je učinkovito.
- Sistemski nedostatak karnitina se manifestuje progresivnom miopatijom, uključujući kardiomiopatiju, i disfunkcijom jetre, praćenu kliničkim simptomima hepatična encefalopatija prema vrsti Reyeovog sindroma. Nedostatak karnitina razlikuje se od potonjeg po ponavljajućem toku i teškoj slabosti mišića koja perzistira između perioda pogoršanja encefalopatije. Nivoi kreatin fosfokinaze u serumu su značajno povišeni, a nivoi karnitina smanjeni i u serumu i u mišićima. Promjene u uzorku biopsije slične su onima s nedostatkom karnitina u mišićnom tkivu. Slične kliničke i morfološke promjene, uključujući nedostatak karnitina, može se otkriti u slučajevima poremećenog metabolizma organskih kiselina, na primjer, s metilmalonskom i glutarnom acidurijom (sekundarni nedostatak karnitina).
Rice. 21-6. Dijete sa urođenim odsustvom lijevog majora prsnog mišića.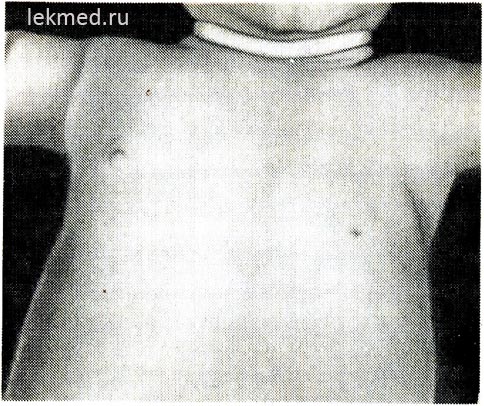
Važno je napomenuti odsustvo prednjeg aksilarnog nabora i nisko ležeće bradavice.
Liječenje se sastoji u tome da pacijent slijedi dijetu bogatu ugljikohidratima i malo masti, te uzimanje karnitina u dnevna doza 100 mg/kg.
Urođeni defekti mišića. Kongenitalno odsustvo mišića. Nerazvijenost mišića može biti prilično česta i dovesti do potpune blokade pokreta zgloba ili kongenitalne artrogripoze. Kao urođena mana najčešće nedostaje jedan mišić. Prilično česta anomalija je odsustvo sternualnog dijela velikog prsnog mišića (sl. 21-6), u nekim slučajevima ovaj defekt je u kombinaciji sa sindaktilijom na zahvaćenoj strani (Poland sindrom). Odsustvo prsnog mišića često prati mišićnu distrofiju. Kongenitalno odsustvo trbušnih mišića abdomen je često povezan s razvojnim defektima urinarnog trakta.
Rice. 21-7. Deformitet vrata i asimetrija lica kod dječaka s kongenitalnim tortikolisom, neliječen od 12. godine. 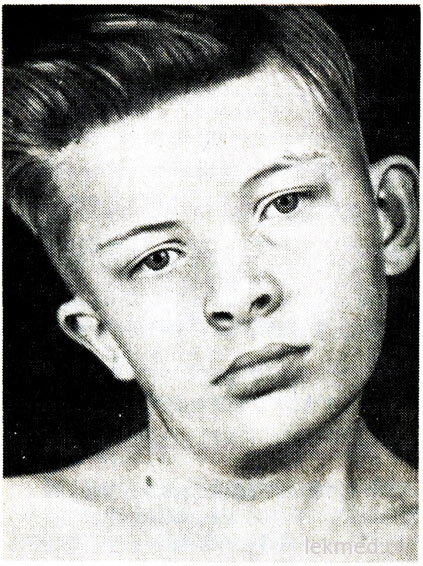
Kongenitalni tortikolis je uzrokovan jednostranim skraćivanjem ili kontrakturom sternokleidomastoidnog mišića. Glava pacijenta je nagnuta prema kontrakturi, a brada je usmjerena prema dolje u suprotnom smjeru (sl. 21-7). Prilikom pokušaja ispravljanja položaja glave osjeća se značajan otpor mišića. U zahvaćenom mišiću se osjećaju zbijena područja. Uzrok kvara je nejasan, dugo se smatralo da je posljedica porođajna trauma. Međutim, tortikolis se javlja kod djece rođene operacijom carski rez; ovo sugerira da se u nekim slučajevima uzrok defekta odnosi na prenatalni period. Tortikolis treba razlikovati od patološkog nagiba glave zbog deformacije vratnih pršljenova, na primjer, s Klippel-Weil anomalijom, te od prijeloma ili dislokacija vratnih kralježaka. Isključuju se radiografskim pregledom. Kod starije djece može doći do naginjanja glave uz strabizam, distoniju, tumore stražnjeg lobanjske jame I cervikalna regija kičmena moždina, miozitis ossificans, cervikalni limfadenitis ili dijafragmalna kila. U većini slučajeva kongenitalni tortikolis može se korigovati pomoću terapijske vježbe. Međutim, kada hronični oblik tortikolis dovodi do asimetričnog razvoja lica i glave (vidi sliku 21-7), što može zahtijevati disekciju mišića iz kozmetičkih razloga.
Kongenitalne miopatije. U ovu grupu spada nekoliko rijetkih oblika nasljednih bolesti kod kojih se javljaju slabost mišića i hipotenzija djetinjstvo(Vidi tabelu 22-1). Njihova tačna dijagnoza ima veliki značaj sa stanovišta prognoze. Općenito je povoljan za normalno funkcioniranje i očekivani životni vijek, za razliku od Werdnig-Hoffmannove bolesti ili kongenitalne mišićne distrofije. Otkrivanje kongenitalnih miopatija obično je olakšano biopsijom mišića.
- Bolest centralnog jezgra. Središnji dio mišićnih vlakana je nenormalno, ali ujednačeno obojen. Elektronski mikroskopski pregled otkriva smanjenje broja mitohondrija i iscrpljivanje sarkoplazmatskog retikuluma u središnjem dijelu vlakana.
Nemalinska miopatija. Pojam "nemalin" objašnjava se činjenicom da su strukture slične filamentima definirane u mišićnim vlaknima.
Rice. 21-8. Miotonična kontrakcija jezika (a) oštrim udarcem udarnim čekićem po desnoj polovini i očnim kapcima (b) kod djeteta sa hiperkalemičnim oblikom porodične periodične paralize. 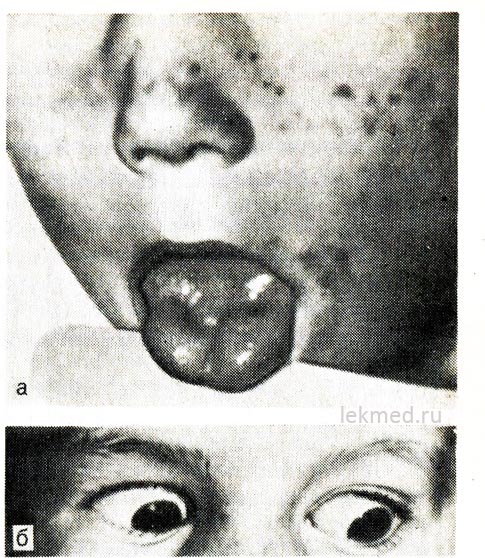
Kada pogledate prema dolje, kapak ostaje skupljen.
Elektronski mikroskopski podaci ukazuju da je to rezultat promjena u Z-trakama miofibrila.
Mitohondrijalne miopatije. Zabilježeni su neki oblici miopatija kod kojih se najvažnije promjene događaju u mitohondrijima mišićnih vlakana. Mogu se značajno povećati i u broju i u veličini. Slabost mišića i hipotenzija mogu se otkriti već u ranom djetinjstvu, ali ponekad primjetno napreduju tek u školskoj dobi. Kardiomiopatija, encefalopatija i laktacidemija često prate miopatije ove grupe.
Miotonija. Ovo stanje je znak različitih bolesti mišića, kao što su distrofična miotonija, hiperkalemični oblik porodične paroksizmalne paralize i bolesti skladištenja glikogena. Miotonija se definira kao značajno kašnjenje u opuštanju mišića nakon voljnih ili prisilnih kontrakcija. Klinički se manifestuje u nemogućnosti otpuštanja šake ili u vidljivoj produženoj kontrakciji mišića nakon njihove stimulacije, izraženoj u oštroj iritaciji (Sl. 21-8). To se može primijetiti ako udarnim čekićem udarite površnu mišićnu grupu, na primjer, mišiće jezika ili palmarnu površinu u području eminencije prvog prsta. Miotonija je potvrđena elektromiografskim podacima. U ovom slučaju, karakteristična spontana mišićna aktivnost je uočljiva nakon opuštanja ili dobrovoljne kontrakcije (miotonična pražnjenja).
Kongenitalna miotonija (Thomsenova bolest). Jedini znak ove bolesti, koja se nasljeđuje prema dominantnom tipu, je miotonija. Može se manifestirati u djetinjstvu u obliku usporavanja pokreta gutanja i naknadnog povraćanja.
rezultat nemogućnosti normalnog opuštanja mišića ždrijela. U starijem djetinjstvu miotonija se manifestira kao pacijentova nesposobnost da otpusti prste stisnute u šaku. Prilikom prvog pokušaja da napravi neku vrstu pokreta, djetetovi mišići postaju tvrdi. Kada ponavljaju isti pokret više puta, oni se donekle opuštaju. Na primjer, bolesno dijete doživljava velike poteškoće u početku čina hodanja. Prvih nekoliko koraka obično čini vrlo neodlučno i sporo. Nakon nekoliko sekundi hod postaje normalan ili gotovo normalan. Simptomi miotonije pogoršavaju se s nepovoljnim emocionalno stanje strpljiv i rashlađuje tijelo. Mišićna snaga ostaje normalna, mišići su dovoljno razvijeni i često primjetno uvećani, što stvara lažan dojam atletske građe pacijenta.
Dijagnoza se zasniva na kliničkim podacima i podacima elektromiografije. Aktivnost enzima u serumu je u granicama normale. Jedini histološka karakteristika služi kao hipertrofija mišićnih vlakana.
Bolest se razlikuje od distrofične miotonije po odsustvu mišićne slabosti i atrofije i distrofičnih promjena u biopsiji mišićnog tkiva. Liječenje novokainom ili kinidin sulfatom je praćeno efektom i indicirano je za funkcionalne poremećaje. Tijek bolesti je obično benigni, a stanje bolesnika se može poboljšati s godinama.
Paroksizmalna paraliza. Ovu grupu bolesti karakterizira periodična slabost mišića uz potpunu ili gotovo potpunu obnovu mišićne snage u periodu između napada. Ovo također uključuje nedostatak mišićne fosforilaze (McArdle bolest).
Hiperkalemijska paroksizmalna paraliza. Nasljedna epizodna adinamija, ili paramiotonija, prenosi se prema dominantnom tipu i posebno je teška kod muškaraca. Obično počinje u ranom djetinjstvu (ponekad u djetinjstvu). Napadi se javljaju tokom perioda odmora nakon teškog mišićnog napora. Slabost se brzo razvija i može trajati nekoliko sati. Posebno se osjeća u nogama; respiratorna funkcija obično nije narušena. Često je adinamija praćena miotonijom, koja perzistira između napada, što se najjasnije manifestira u obliku kašnjenja u kretanju očnih kapaka kada se gleda prema dolje (vidi sliku 21-8, b).
Nivoi kalijuma u serumu su često povišeni tokom napada, ali može biti potrebno ponovljeno testiranje tokom više napada kako bi se to pouzdano utvrdilo. Napad se može vještački isprovocirati upotrebom kalija (2-3 g oralno), ali ga treba izvoditi samo uz EKG praćenje. Ponovljeni napadi se zaustavljaju dijakarbom. Teške oblike bolesti karakterizira razvoj kronične, blage slabosti i distrofičnih promjena u mišićima.
Hipokalemijska paroksizmalna paraliza. Porodična paroksizmalna paraliza, također dominantno naslijeđena, posebno je teška kod dječaka. Za razliku od hiperkalemične forme, prvi napad se javlja u kasnom ili ranom djetinjstvu. adolescencija. Uzrok je konzumiranje obilnog obroka bogatog ugljikohidratima ili odmor nakon fizičke aktivnosti. Obično napad počinje sljedećeg jutra nakon teške fizičke aktivnosti i obilne večere. Karakterizira ga slabost mišića i arefleksija. Funkcija disanja može biti oštećena. Može doći do aritmije, uključujući ventrikularne ekstrasistole i tahikardiju. Napadi mogu trajati više od 24 sata.U paralitičkoj fazi obično se smanjuje nivo kalijuma u serumu (2-3 mmol/l). Osnovni defekt je nepoznat. Pacijenti s ponovljenim teškim napadima razvijaju kroničnu slabost mišića i patoloških promjena u mišićima. Liječenje tokom napada se sastoji od uzimanja kalijum hlorida; njegova početna doza je 2-3 g. Diakarb pomaže u smanjenju učestalosti napada.
Paroksizmalna mioglobinurija (idiopatska mioglobinurija). Idiopatska mioglobinurija je heterogena grupa bolesti kod kojih se napadi paralize s mioglobinurijom javljaju spontano ili nakon intenzivne fizičke aktivnosti. Bolest se nasljeđuje na dominantan način, vezana za X hromozom. Mišići, najčešće mišići potkoljenice i butine, postaju bolni i otečeni tokom napada. Urin postaje tamnocrven ili Smeđa boja. Mioglobinurija može uzrokovati nekrozu bubrežnih tubula, što dovodi do smrti zatajenje bubrega.
Dijagnoza se potvrđuje otkrivanjem mioglobulina u urinu. Pozitivan benzidinski test u odsustvu crvenih krvnih zrnaca u urinu potvrđuje prisustvo mioglobina u njemu, posebno ako hemoglobin nije otkriven u serumu. Hemoglobin se određuje spektrofotometrijom. Paroksizmalnu mioglobinuriju treba razlikovati od McArdleove bolesti, nedostatka karnitin palmitil transferaze i mioglobinurije nakon neuobičajeno intenzivne fizičke aktivnosti ili traume mišića u zdrava osoba. Mioglobinurija nakon teške mišićne vježbe javlja se kod pseudohipertrofične mišićne distrofije (Duchenneova bolest).
Tretman se sastoji od mirovanja u krevetu; Ako je potrebno, izvršite umjetnu ventilaciju. Da bi se spriječilo zatajenje bubrega, potrebno je pacijentu prepisati dosta tekućine.
Nedostatak karnitin palmitil transferaze. Kada je ovaj enzim nedovoljan, prijenos dugolančanih masnih kiselina u mitohondrijalne segmente, u kojima dolazi do oksidacije i proizvodnje ketona, je poremećen. Nedostatak izoenzima tipa II se nasljeđuje na recesivan način. Zbog njegovog nedostatka, ketogeneza u tkivima, uključujući mišiće i jetru, je poremećena. Prvi znaci bolesti češće se javljaju kod djece školskog uzrasta i tinejdžera. Sastoje se od ponavljanih epizoda bolova u mišićima, slabosti i groznice nakon vježbanja ili posta. Mioglobinurija koja prati napade može dovesti do zatajenja bubrega. Post dovodi do hipoglikemije. Između napada, djeca izgledaju zdrava. Bolest se mora razlikovati od drugih stanja praćenih periodičnom slabošću i mioglobinurijom. Metoda za određivanje aktivnosti karnitin palmitil transferaze ima diferencijalnu dijagnostičku vrijednost. Smanjuje se u mišićnom i jetrenom tkivu, leukocitima i kulturi fibroblasta. Pridržavanje dijete koja se sastoji od hrane obogaćene ugljikohidratima i hrane s niskim udjelom masti pomaže u smanjenju broja napada.
Mišićne distrofije. Ove anomalije spadaju u grupu porodičnih bolesti praćenih degeneracijom mišićnih vlakana. Klasifikacija mišićnih distrofija zasniva se na karakteristikama kao što su vrijeme nastanka, brzina progresije, distribucija lezija po mišićnim grupama i način nasljeđivanja.
Pseudohipertrofična mišićna distrofija. Djetinjstvo ili Duchenne je najčešći oblik mišićne distrofije; njegova učestalost je 0,14 na 1000 djece. U svom klasičnom obliku, javlja se samo kod dječaka, a X-vezano nasljeđivanje javlja se u približno 50% probanda. U drugim slučajevima, bolest je uzrokovana novim mutacijama. Prijavljen je rijedak oblik mišićne distrofije, klinički identičan Duchenneovom obliku, ali se nasljeđuje na recesivan način sa istom učestalošću bolesti kod dječaka i djevojčica. Rijetko je moguće pouzdano dijagnosticirati bolest kod djeteta mlađeg od 3 godine. Anamneza obično ukazuje da je dijete imalo zastoj u razvoju motoričke funkcije, kasno je počeo sjediti, hodati i trčati, što, naravno, ukazuje na raniju pojavu bolesti. Česti su pačji hod, otežano penjanje uz stepenice, hipertrofija mišića lista kliničke manifestacije. U nekim slučajevima, u proces su uključeni i drugi mišići, posebno mišići deltoida, brahioradialis i jezik.
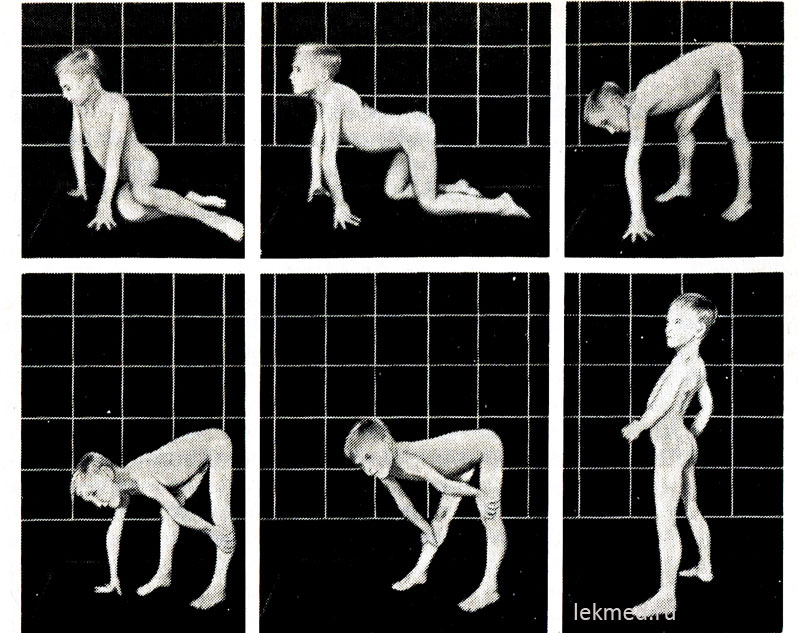
Rice. 21-9. Tipični položaji koje je zauzelo 7-godišnje dijete s pseudohipertrofičnom miopatijom kada ustaje s poda (Gauersov znak).
U stojećem položaju ( zadnja fotografija) postoji značajno izražena lordoza.
Na početku bolesti hipertrofirani mišići imaju značajnu snagu, ali kasnije ona opada (pseudohipertrofija), jer do povećanja mišićne mase dolazi zbog njihove masne infiltracije. Snaga hipertrofiranog mišića lista znatno premašuje snagu mišića prednje površine noge, što objašnjava česte kontrakture petne tetive i hodanje djeteta na prstima. Slabost mišića karličnog pojasa izražava se karakterističnim pačjim (gospodarskim) hodom i poteškoćama koje dijete doživljava pri ustajanju iz sedećeg položaja na podu. Kad dosta teški oblici mišićna distrofija kod djeteta, primjećuje se Goversov simptom: pri ustajanju s poda prvo kleči, oslanjajući se na ruke, a zatim se diže, uzastopno rukama odgurujući potkoljenice, zglobovi kolena i bokova (sl. 21-9). Slabost mišića ramenog obruča može se utvrditi držanjem djeteta u povišenom položaju za pazuhe. Obično pokušava da se izdrži pritiskajući ruke uz telo; sa mišićnom distrofijom, čini se da izmiče iz ruku ispitivaču. Bolesno dijete često ne može podići ruke iznad glave. IN kasne faze Bolest razvija značajnu atrofiju mišića. Tipično, do 12. godine dijete više ne može hodati. Pacijenti u 75% slučajeva umiru prije 20. godine života. Većina njih ima kardiomiopatiju, koja u nekim slučajevima uzrokuje iznenadnu smrt. Ako je nasljeđe povezano s X hromozomom, a bolest je počela u kasnom djetinjstvu, očekivani životni vijek ostaje dug (Beckerova mišićna distrofija). Prosječni koeficijent mentalni razvoj kod djece sa Duchenneovom formom je 80; 25% djece ima mentalnu retardaciju.
At diferencijalna dijagnoza Duchenneovu mišićnu distrofiju treba uzeti u obzir kod Werdnig-Hoffmannove bolesti kod starije djece i bolesti mišića kao što su endokrine miopatije, nedostatak karnitina, bolesti skladištenja glikogena i polimiozitis. Ponekad se kod kontraktura petne tetive i hodanja djeteta na prstima može pretpostaviti cerebralna paraliza, ali kod mišićne distrofije nema karakterističnih znakova. cerebralna paraliza spastičnost i hiperrefleksija.
Dijagnoza se zasniva na određivanju aktivnosti enzima u serumu, podacima elektromiografije i biopsiji mišićnog tkiva. Aktivnost enzima, posebno kreatin fosfokinaze, čak i prije razvoja kliničkih simptomačesto premašuje normu 10 puta čak i kod dojenčadi. Elektromiogram prvenstveno otkriva smanjenje trajanja i smanjenje amplitude motoričkih potencijala. Histološke promjene se sastoje od degeneracije mišićnih vlakana. Često se razlikuju po veličini i djelimično su zamijenjeni masnim i vezivnim tkivom. Veličina njihovih jezgara također varira. Dijagnoza se može postaviti pri rođenju mjerenjem aktivnosti kreatin fosfokinaze. Metode za identifikaciju ženskih nositelja još uvijek nisu razvijene, unatoč činjenici da 60-80% njih ima blagi ili umjereni porast njegovog nivoa. Ovi znakovi su tipičniji za djetinjstvo nego za naredne periode života.
Ne postoje efikasni tretmani. Pacijent treba da bude aktivan i da može hodati što je više moguće. Potrebno je osigurati da dijete izbjegava intenzivnu fizičku aktivnost, jer to može uzrokovati pucanje mišićnih vlakana. U nekim slučajevima, hirurško produženje petne tetive poboljšava sposobnost hodanja, ali se produžava odmor u krevetu nakon ortopedske korekcije može povećati atrofiju mišića. Genetsko savjetovanje igra važnu ulogu.
Kongenitalna mišićna distrofija. Bolest se nasljeđuje autosomno recesivno i karakterizira je mišićna hipotonija i slabost kod novorođenčadi. Uvršten je u grupu stanja koja su definisana kao „mlohavo dete“ (videti tabelu 21-1). Početak bolesti datira iz prenatalnog perioda. Ponekad novorođenče ima tešku atrofiju mišića, kontrakture i ograničenu pokretljivost zglobova. Diferencijacija od Werdnig-Hoffmannove bolesti je teška. Fascikulacije jezika, karakteristične za potonje, izostaju kod mišićne distrofije. Tetivni refleksi su depresivni, ali nisu potpuno izgubljeni. Proces uključuje mišiće uključene u disanje, uključujući dijafragmu. U teškim slučajevima, smrt se javlja prije navršene 1 godine života zbog respiratorne insuficijencije; u blažim oblicima, normalna održivost traje dugo vremena. Nije zabilježeno povećanje aktivnosti serumskih enzima, iako se javljaju distrofične promjene u mišićima.
Facijalno-humeralni oblik mišićne distrofije. Ovo je dovoljno lagana forma mišićna distrofija se nasljeđuje autosomno dominantno. Obično počinje u dobi od 10-20 godina i karakterizira ga slabost i atrofija mišića lica i ramenog pojasa. Lice je potpuno prijateljsko, pacijent ne može sklopiti oči i zviždati. Bolest napreduje sporo i kompatibilna je sa normalno trajanježivot. Dijagnoza se zasniva na kliničkim nalazima i načinu nasljeđivanja. Rezultati biopsije mišićnog tkiva ukazuju na distrofične promjene u njemu. Nivoi kreatin fosfokinaze u serumu mogu ostati u granicama normale ili mogu biti blago povišeni.
Karlični oblik mišićne distrofije. Ovu grupu heterogenih poremećaja karakterizira sporo napredovanje mišićne distrofije i nasljeđuje se autosomno recesivno. Početak bolesti odnosi se na starije djetinjstvo, adolescenciju ili odraslu dob. Obično su zahvaćeni mišići karličnog pojasa.
Oblik očiju miopatije. Distrofične promjene se javljaju uglavnom u vanjskom dijelu očne mišiće. Bolest počinje u djetinjstvu ili adolescenciji. Sa njim napreduje ptoza i ograničenje pokreta očne jabučice. Ponekad se slabost širi na mišiće lica i vrata. Bolest treba razlikovati od mijastenije gravis i paralize kranijalni nervi za tumore moždanog stabla.
Progresivna oftalmoplegija, koja počinje u djetinjstvu ili adolescenciji, povezana je s atipičnom pigmentnom degeneracijom retine i srčanim blokom (Kearns-Sayersov sindrom). Obično je povezan s progresivnom ataksijom, usporavanjem rasta i pubertetom. Ispod mišićne sarkoleme otkrivaju se velike nakupine atipičnih mitohondrija. Genetska priroda ovog procesa nije utvrđena. Mogućnost iznenadne smrti od problema s provodljivošću srca može se kontrolirati korištenjem pejsmejkera.
Miotonična distrofija. Unatoč činjenici da miotonična distrofija počinje kao kod odrasle osobe, njen početak se sve češće bilježi kod dojenčadi i kasnijeg djetinjstva. Nasljeđuje se autosomno dominantno. Njegov početak u djetinjstvu ukazuje da majka pati od miotonije. Shodno tome, intrauterini faktori mogu uticati na težinu bolesti kod deteta. Već u trenutku rođenja može imati mišićnu hipotoniju, a nedostaje mu sposobnost sisanja. Usporen fizički i mentalni razvoj obično se otkrije kasnije. U ranom djetinjstvu, mišićna slabost i atrofija širi se uglavnom na mišiće lica, vilice i temporalne mišiće. Obično se primjećuje bilateralna ptoza. K dijagnostički smislene metode uključuju perkusiju mišića, elektromiografiju; tipična za ove pacijente je nemogućnost otpuštanja ruke stisnute u šaku (vidi Kongenitalna miotonija). Slabost i atrofija mišića udova i karličnog pojasa (obično distalne grupe) otkrivaju se u starijem djetinjstvu ili adolescenciji. Kod odraslih ovu bolest prati katarakta, ćelavost i atrofija testisa.
Dijagnoza se zasniva na identifikaciji znakova miotonije, karakteristične distribucije mišićne slabosti, dominantnog nasljeđa i distrofičnih promjena u mišićima. U djetinjstvu tok bolesti može biti nepovoljan, često praćen mentalnom retardacijom. TO adolescencija mišićna slabost dolazi do izražaja. Za funkcionalne poremećaje indicirano je liječenje novokainom i kinidinom.
Upala je vrlo bolna bolest koju karakteriziraju upalni procesi u skeletnih mišića, With iz raznih razloga pojava. IN medicinska praksa naziva se miozitis. Glavna karakteristika je opšta slabost mišića, što je izazvano upalnim procesima u mišićno tkivo, odgovoran za opterećenje motora. Ova upala također uključuje druge vrste povezane s infekcijama, toksinima i često štetnim efektima. lijekovi. Glavni upalni procesi su višestruki i povezani s lezijama površne prirode.
Ko češće dobija miozitis?
Prema dugogodišnjim zapažanjima, smatra se da je učestalost takve bolesti od dva do deset slučajeva na milion prosječne ljudske populacije. Bolest karakteriziraju dva vrhunca pojavljivanja vezana za starost. Prvi se javlja u dobi od pet do šesnaest godina. Drugi vrh se javlja na više senior grupa od četrdeset do šezdeset godina. Utvrđen je i rodni prioritet u bolesti, žene su više pogođene ovom bolešću, odnos oboljelih žena i oboljelih muškaraca varira u prosjeku od dva do tri kod žena i jedan kod muškaraca.
Koji su uzroci upale?
Tačni uzroci pojave još nisu razjašnjeni ove bolesti. Ali sezonski uticaj, u smislu učestalosti pojavljivanja, ne ukazuje direktno na povećanje broja bolesti u zimski period ili početkom proljeća, a to je upravo vrhunac prekoračenja pragova epidemije zarazne bolesti. Ako uzmemo u obzir genetski utjecaj na pojavu ovih upala, onda se one uglavnom manifestiraju kod identičnih blizanaca i direktnih srodnika koji su već oboljeli od ove bolesti. Takvo prenošenje genetskih informacija obično nije povezano sa samom bolešću, već je povezano sa specifičnim poremećajima u imunološkom sistemu.
Prvi znaci miozitisa
Glavni pokazatelji toka bolesti su poremećaji imunološkog odgovora na ćelijski nivo. Prilikom imunopatološkog istraživanja upaljene mišićne površine, otkriva se patološka infiltracija nekih ćelijskih tijela koja su u početnom aktivnom stanju i oslobađa se toksin koji utiče na procese oporavka. Uz kožnu manifestaciju bolesti moguće je identificirati određene patogene koji remete ukupnu ćelijsku rezistenciju. Otkriva se i reakcija antitijela, koja u nekim slučajevima ima negativne reakcije na krvnim sudovima.
U početnom toku bolesti česte su posjete pacijenata zbog slabosti i velike slabosti i upalnih procesa na koži. Tokom vremena, nekoliko sedmica, dolazi do povećanja početnih znakova i povećanja slabosti i, motoričkih mišića ah ruke i stopala. U slučaju bolesti u mladom tijelu, uočavaju se akutni početni simptomi, praćeni izraženim promjenama u konstituciji tijela, bilo gubitkom težine, bilo grozničavim pojavama.
Ako pogledamo starije pacijente, oni doživljavaju postupno povećanje mišićne slabosti, ponekad tokom nekoliko godina. Površinske lezije kože. Neki pacijenti također mogu doživjeti specifični znakovi, koje su praćene vaskularnim grčevima i poremećajima cirkulacije, poremećajima u koštanog tkiva i otežano disanje, koje nastaje kao posljedica određenih upalnih procesa u plućima.
Kako se bolest manifestuje?
Prva manifestacija ove bolesti je klinička tačka vid je opća slabost svih mišićnih grupa odgovornih za motoričku aktivnost, kako gornjih tako i niže grupe ovi mišići, kao i vratna kičma. Ovi poremećaji dovode do toga da pacijent u početnoj fazi ima poteškoće u kretanju nakon dužeg perioda mirovanja, otežano ustajanje iz sjedećeg položaja ili ulazak ili izlazak iz vozila ili u drugim svakodnevnim pokretima. Izvana su vidljive smetnje u hodu, postaje nejasan, ne mnogo nekoordiniran.
Postoje slučajevi kada pacijent ne može sam ustati iz kreveta. Upala mišića također može zahvatiti područje povezano s gutanjem i glasom, što može biti praćeno akutnim poteškoćama pri pokušaju gutanja, koje završavaju eksplozivnim kašljem. Takođe, kod nekih pacijenata se javlja dermatomiozitis, koji se manifestuje u vidu područja pojačanog osipa na pojedinim delovima tela ili na licu ili vratu i drugim delovima tela. Ima i drugih kožne manifestacije, koji direktno upućuju na tok bolesti, mogu se kombinirati i s manifestacijama polimiozitisa, to mogu biti ljuskavi i crvenilo kože ili čak popraćeno mikropukotinama na koži u nekim područjima gornjih ekstremiteta.
Taloženje kalcijevih soli u mekim tkivima
Taloženje soli u mekim tkivima (kalcifikacija) je obično proizvod dugotrajne bolesti, kasnijim fazama upalnih procesa. Formacije se pojavljuju i obično su lokalizirane ispod kože ili na mjestima mišića koji spajaju tkivo, iznad zglobnih dijelova koljena i laktova, iznad falangi prstiju i mnogih drugih, pa čak i na nekim područjima stražnjice.
Upalni procesi u plućima s miozitisom
Jasan inicijalni znak upalnih procesa u plućima tokom miozitisa je respiratorna otežano disanje, obično je povezana s disfunkcijom mišića dijafragme ili kvar srca ili s upalom povezane s toksinima sadržanim u lijekovima koje pacijent uzima. Upalni procesi su takođe praćeni kašljem ili insuficijencijom respiratorna aktivnost. Posebno teški slučajevi Može se razviti zagušljiva pneumonija.
Kako miozitis utiče na srce?
Upalni procesi koji se javljaju u srčanom mišiću tijekom miozitisa obično se javljaju bez izraženih simptoma. Samo kada specijalno istraživanje Uz pomoć kardiograma uočavaju se neki poremećaji u ritmovima otkucaja, koji se obično pripisuju područjima drugih bolesti. Zatajenje srca se praktički ne opaža.
Dijagnoza upale laboratorijskim pretragama
Prilikom provođenja krvnih pretraga nisu uočene ozbiljne promjene. Ostali testovi otkrivaju odstupanja koja su karakteristična za poremećaje mišića odgovornih za motoričku aktivnost (CPK). U nekim tokovima bolesti, oni se primjećuju kod gotovo svih pacijenata. Zasićenost ovog indikatora može se povećati sve dok se ne pojave očigledni početni znakovi klinički pregled. Ali ponekad ovaj pokazatelj može biti u granicama normale i čak ne ukazuje na tešku upalu mišićnog tkiva. Postoji i indikator niske motoričke sposobnosti koji ukazuje na neke moguće opšti simptomi sa hepatitisom (transminaze).
Liječenje upale mišića
U praksi otkrivaju. Da je upala mišića bolest koja se javlja periodično, odnosno s egzacerbacijama. Bol koji se javlja kod drugih hronične bolesti su stalne, au slučaju ove bolesti su periodične i ne smetaju stalno. Miozitis ima dva stadijuma bolesti: hronični i akutni. Uz pravilan tretman, akutna faza bolesti glatko prelazi u kroničnu, manje bolnu. U hroničnom stadijumu bolesti upalni procesi postaju osjetljivi na sezonske promjene ili klimatska promjena. Hronična upala može biti uzrokovan ne samo kao stadij nakon akutne faze miozitisa. Ali i za neke zarazne bolesti. Stoga često ljudi čak i ne sumnjaju na miozitis kada su bolesni od uobičajene zarazne gripe, ali nakon oporavka pojavljuju se znakovi oštećenja mišića. Akutna upala mišići se mogu manifestirati na lokalnoj lokaciji.
Vrlo je teško otkriti bolest miozitis, jer je striktno periodična. Ali kod prvih simptoma potrebno je kontaktirati stručnjaka. To je potrebno učiniti iz više razloga, ali prvi bi trebao biti postavljanje tačne dijagnoze, a nakon toga propisivanje optimalan tretman, glavna stvar je da je sistemske prirode.
Liječenje upale mišića može uključivati pravilnu raspodjelu opterećenja na tijelo s fizičkim utjecajem na bolest. U nekim slučajevima se koriste sportske aktivnosti koje se izmjenjuju s periodima potpunog opuštanja. Primarni zadatak u liječenju miozitisa je utvrđivanje uzroka upale. Na osnovu istraživanja, oni propisuju liječenje lijekovima uz pomoć specijalizovanih lekova. Obično se za liječenje propisuje skup lijekova, ali svaki sistem uključuje lijekove protiv bolova na bazi analgetika i lijekove koji sprječavaju upalne procese. Koriste se mnogi lijekovi, ali glavni su Diklofenak, Ketonal, Nurofen.
Za lokalna ili lokalizirana žarišta bolesti koriste se posebni lijekovi koji se propisuju u obliku masti za zagrijavanje: Apisatron, Nicoflex, Finalgon. Pri liječenju upalnih procesa kod djece obično se propisuju specijalizirani lijekovi sa smanjenom dozom komponenti za liječenje, uključujući u ovom slučaju Tu je dobre povratne informacije o drogi Doktor MOM.
U periodima kada se tok bolesti pogoršava, ni u kom slučaju ne treba preopteretiti organizam. fizička aktivnost i općenito morate se pridržavati odmora u krevetu. Ovo je od primarne važnosti kod upalnih procesa u području kičmenih motornih mišića. Kod takve egzacerbacije potrebno je liječenje sledeće lekove lijekovi protiv bolova i lijekovi koji sprječavaju upalne procese, lista takvih lijekova uključuje: Brufen, Reopirin, Indometacin.
Takav tretman može biti efikasan samo ako Ako je sistemski i ako se koristi u kombinaciji s drugim efikasne metode i sredstva. Iz ovoga proizilazi da se pored uzimanja lijekova moraju provoditi i fizioterapeutski postupci i vrlo je poželjno da se koriste vježbe koje su uključene u terapijsko-preventivno fizičko vaspitanje. Ali treba imati na umu da je u slučaju akutnog miozitisa motornih mišića kralježnice takva fizička vježba strogo kontraindicirana. Prilikom povećanja opšta temperatura u toku bolesti potrebno je koristiti lekove koji je smanjuju i obavezno ograničiti kontakt sa hladnom atmosferom.
U slučajevima kada su upalni procesi gornjih mišića, odgovornih za motoričko opterećenje na vratu, uzrokovani septičkim lezijama, tada je nakon identifikacije potrebno kontaktirati praktičarskog kirurga, koji je u obavezi da ovo područje kirurški otvori i ukloni. svu infekciju. Također, kod ove vrste upale mišića strogo je zabranjena svaka vrsta masaže.
Svaka vrsta miozitisa ima svoje karakteristike koje su jedinstvene za njega. Na primjer, upalni proces cervikalne mišićne grupe može se liječiti bez posebnih komplikacija i relativno je lako ako se s tim liječenjem ne započne u uznapredovaloj fazi bolesti, već odmah kada se pojave prvi znaci. Prilikom liječenja ove vrste miozitisa, stručnjaci obično propisuju sjedeći tretman ili, što je najbolje, mirovanje u krevetu. Specijalisti za liječenje propisuju kompleks lijekova, uključujući masti za zagrijavanje, koje se utrljaju u područje upale i uzimaju lijekove koji sprječavaju upalne procese.
U praksi, takozvana “novokainska blokada” daje odlične rezultate. Proces ovog događaja uključuje ciljanu primjenu anestetika oko upaljenog područja s novokainom i posebnim hormonom. Ali daje pozitivan efekat, samo u slučajevima kada ih pacijent nema alergijske reakcije i kontraindikacije. Postoji i metoda istezanja mišićnog i ligamentnog tkiva za postupak opuštanja. Ovo je apsolutno nova metoda, koji daje odlične rezultate i već je testiran u mnogim medicinskim institutima.
U priloženom videu možete otkriti zašto se tetive upale.
Ali najjednostavnije preporuke koje prate svakog potencijalnog pacijenta dobar specijalista, su preporuke da se kritična područja mišića zaštite od hladnoće, da ne stagniraju i da se ne zadržavaju dugo u jednom statičkom položaju, kako u ležećem tako iu sjedećem položaju. Birajte samo udobne poze u kojima se mišići ne ukoče, izbjegavajte propuh i izvodite opće vježbe jačanja. Ove jednostavne i prilično nekomplicirane preporuke pomoći će vam da se nepotrebno ne izlažete upalnim procesima i miozitisu.