Οι νευρομυϊκές παθήσεις χαρακτηρίζονται από διαταραχή της λειτουργίας των εκούσιων μυών, απώλεια ή μείωση του κινητικού ελέγχου, η οποία μπορεί να προκύψει ως αποτέλεσμα βλάβης και στους ίδιους τους μύες και να είναι δευτερεύουσας φύσης - λόγω δυσλειτουργίας της νευρομυϊκής σύνδεσης, βλάβη περιφερικά νεύραή κινητικούς νευρώνες νωτιαίος μυελός. Στην κλινική εικόνα ορισμένων νευρομυϊκών παθήσεων μπορεί να υπάρχουν σημεία βλάβης στους κινητικούς πυρήνες του εγκεφαλικού στελέχους. Βλάβες σε άλλες περιοχές νευρικό σύστημα, που οδηγεί σε παραβίαση του κινητικού ελέγχου, ιδίως της πυραμιδικής οδού, σύμφωνα με τον γενικά αποδεκτό ορισμό, δεν ανήκουν σε νευρομυϊκές παθήσεις.
Τα πιο κοινά συμπτώματα νευρομυϊκών παθήσεων είναι αδυναμία, μειωμένος μυϊκός όγκος (ατροφία), ακούσιες μυϊκές συσπάσεις, σπασμοί, μούδιασμα, μυρμήγκιασμα κ.λπ. Μια δυσλειτουργία της νευρομυϊκής συμβολής μπορεί να προκαλέσει πεσμένα βλέφαρα (πτώση), διπλή όραση (διπλωπία) και άλλα σημάδια μυϊκής αδυναμίας που αυξάνονται κατά τη διάρκεια της ημέρας. Σε ορισμένες ασθένειες, η κατάποση, ακόμη και η αναπνοή, μπορεί να διαταραχθεί.
Μυϊκές παθήσεις: Συμπτώματα
- προοδευτικές μυϊκές δυστροφίες– γενετική κληρονομική ασθένειαμύες, τα συμπτώματα των οποίων εμφανίζονται συνήθως στη βρεφική ηλικία ή Παιδική ηλικίαλιγότερο συχνά στους ενήλικες. Η μυϊκή αδυναμία αυξάνεται σταδιακά, ιδιαίτερα αισθητή σε αυθαίρετους μύες. Αυτή η ομάδα περιλαμβάνει μυϊκή δυστροφία Becker, συγγενή μυϊκή δυστροφία, περιφερική μυϊκή δυστροφία, μυϊκή δυστροφία Duchenne (η πιο κοινή μορφή μυϊκής δυστροφίας στα παιδιά), μυϊκή δυστροφία Emery-Dreyfus, μυϊκή δυστροφία ώμου-ωμοπλάτης, μυοτονική μυϊκή δυστροφία (η πιο κοινή μυϊκής δυστροφίας σε ενήλικες), οφθαλμοφαρυγγική μυοδυστροφία.
- Φλεγμονώδεις μυοπάθειες- ονομάζεται επίσης μυοσίτιδα, βασίζεται σε μια φλεγμονώδη διαδικασία που οδηγεί σε μυϊκή αδυναμία, ο ρόλος των αυτοάνοσων διαταραχών τονίζεται στην ανάπτυξή τους, μερικές φορές σε συνδυασμό με άλλες αυτοάνοσο νόσημα. Αυτές περιλαμβάνουν δερματομυοσίτιδα, πολυμυοσίτιδα, μυοσίτιδα με εγκλείσματα.
- Μιτοχονδριακές μυοπάθειες- προκύπτουν ως αποτέλεσμα δομικών ή βιοχημικών ελαττωμάτων στα μιτοχόνδρια. Σύνδρομο Kearns-Syre, μυοκλονική επιληψία με «σκισμένες κόκκινες ίνες», μιτοχονδριακή εγκεφαλομυοπάθεια.
- Μυοτονία - συγγενής μυοτονία ή νόσος του Thomsen, δυστροφική μυοτονία, συγγενής παραμυοτονία, νευρομυοτονία ή νόσος Isaacs
- Άλλες μυοπάθειες – νόσος του κεντρικού πυρήνα, μυοσωληναριακή μυοπάθεια, μυοπάθεια νεμαλίνης, υπερκαλιαιμική και υποκαλιαιμική περιοδική παράλυση, ενδοκρινικές μυοπάθειες
Παθήσεις της νευρομυϊκής σύνδεσης
Προκαλούν δυσλειτουργία της φυσιολογικής συναπτικής μετάδοσης των παλμών από τις νευρικές απολήξεις στις μυϊκές ίνες. Η ασθένεια μπορεί να βασίζεται σε μια αυτοάνοση διαδικασία.
- βαρεία μυασθένεια
- Σύνδρομο Lambert-Eaton
- συγγενές μυασθενικό σύνδρομο
Παθήσεις των περιφερικών νεύρων
- Μονονευροπάθειες - βλάβη σε ένα νεύρο, η πιο κοινή αιτία είναι ένα φαινόμενο συμπίεσης ( σύνδρομα τούνελ), τραυματικές κακώσεις
- Πολλαπλές μονονευροπάθειες - πολυεστιακές βλάβες των περιφερικών νεύρων, που συνήθως σχετίζονται με συστηματικά ή λοιμώδη νοσήματα, παρανεοπλασματικά σύνδρομα
- Οι πολυνευροπάθειες είναι διάχυτες, συμμετρικές βλάβες των περιφερικών νεύρων, συνήθως επικρατούν στα άπω μέρη, σχεδόν πάντα υπάρχουν διαταραχές ευαισθησίας στην κλινική εικόνα. Μπορεί να είναι οξεία (σύνδρομο Guillain-Barré), χρόνια (χρόνια φλεγμονώδης απομυελινωτική πολυνευροπάθεια), επίκτητη (τοξική, διαβητική, παρανεοπλασματική) ή κληρονομική (περονιαία μυϊκή ατροφία ή νόσος Charcot-Marie-Tous, νόσος Dejerine-Sott, αταξία Friedreich).
- Πλεξοπάθειες - βλάβες των πλεγμάτων των άνω και κάτω άκρων (ώμος και οσφυοϊερός), η πιο κοινή αιτία των οποίων είναι τραυματική ή συμπιεστική πρόσκρουση
- Radiculopathy και polyradiculopathy - βλάβες των κινητικών ή αισθητήριων ριζών της σπονδυλικής στήλης
Ασθένειες κινητικός νευρώνας
Μια προοδευτική εκφυλιστική βλάβη των κινητικών νευρώνων που έχει ως αποτέλεσμα τον εξασθενημένο κινητικό έλεγχο των άνω ή κάτω άκρων, καθώς και διαταραχές του βολβού. Η έναρξη συχνότερα στη μέση ηλικία, τα συμπτώματα μπορεί να περιλαμβάνουν αδυναμία στα άκρα, δυσκολία στην κατάποση, ομιλία, βάδισμα, αδυναμία των μυών του προσώπου και μυϊκούς σπασμούς. Η ομάδα αυτή περιλαμβάνει ειδικότερα:
- πλευρά αμυοτροφική σκλήρυνση(ΜΠΑΣΣΟ)
- νωτιαία μυϊκή ατροφία ενηλίκων
- νωτιαία μυϊκή ατροφία σε βρέφη ή νόσος Werdnig-Hoffmann
- νεανική νωτιαία μυϊκή ατροφία ή νόσος Kugelberg-Welander
- βολβονωτιαία μυϊκή ατροφία ή νόσος Kennedy
Διάγνωσηβασίζεται στο ιστορικό της νόσου, διεξοδική νευρολογική εξέταση, στις περισσότερες περιπτώσεις χρησιμοποιείται ηλεκτρομυογραφική (ΗΜΓ) μελέτη, όταν συνδυάζεται με βλάβη του κεντρικού κινητικού νευρώνα ή για τον αποκλεισμό της, μπορεί να χρησιμοποιηθεί διακρανιακή μαγνητική διέγερση, εάν είναι κληρονομική υπάρχουν υποψίες για μορφές, γίνεται ανάλυση DNA, η αυτοάνοση φύση της διαδικασίας απαιτεί προσδιορισμό ειδικών αντισωμάτων, μπορεί να πραγματοποιηθεί βιοψία μυϊκής περιοχής, με πρωτογενείς μυϊκές βλάβες, ελέγχεται το επίπεδο της κρεατινοφωσφοκινάσης (CPK), σε Πρόσφατακερδίζει επίσης δημοτικότητα υπερηχογραφική εξέτασημύες και περιφερικά νεύρα. Διαγνωστικός αλγόριθμος, επιλογή πρόσθετη έρευναεξαρτώνται από τα χαρακτηριστικά του κλινικού προτύπου και τον εντοπισμό της βλάβης - μυς, νεύρο, πλέγμα, ρίζες, κινητικοί νευρώνες.
ΚΛΗΡΟΝΟΜΙΚΕΣ ΠΑΘΗΣΕΙΣ ΤΟΥ ΝΕΥΡΙΚΟΥ ΣΥΣΤΗΜΑΤΟΣ
ΔΙΑΛΕΞΗ 16
Τα εκφυλιστικά νοσήματα με κυρίαρχη βλάβη του νευρομυϊκού μηχανισμού αποτελούν τη μεγαλύτερη ομάδα όλων των κληρονομικών νοσημάτων.
Εξαιρετικά σημαντικά, και συχνά καθοριστικά στη διάγνωση νευρομυϊκών παθήσεων, είναι τα αποτελέσματα ηλεκτροφυσιολογικών και βιοχημικών μελετών. Η σημασία των παθομορφολογικών ευρημάτων είναι εξίσου μεγάλη. Η μελέτη της μυϊκής βιοψίας σε μικροσκόπιο φωτόςβοηθά στη διαφοροποίηση της μυογενούς ατροφίας από τη νευρογενή ατροφία. Η ιστοχημική εξέταση είναι απαραίτητη για την ταυτοποίηση μεταβολικές βλάβεςμύες, και η ηλεκτρονική μικροσκοπία άνοιξε μια ολόκληρη μεγάλη κατηγορία ασθενειών - μη προοδευτικές μυοπάθειες.
Προοδευτικές μυϊκές δυστροφίες.Ο όρος μυϊκές δυστροφίες είναι μια ομάδα γενετικά καθορισμένων διαταραχών που χαρακτηρίζονται από προοδευτικές εκφυλιστικές αλλαγές στις μυϊκές ίνες χωρίς πρωτογενή παθολογία του περιφερικού (κατώτερου) κινητικού νευρώνα.
Διάφορες μορφέςδιαφέρουν μεταξύ τους ως προς τους τύπους κληρονομικότητας, το χρόνο έναρξης της διαδικασίας, τη φύση και την ταχύτητα της πορείας της, την ιδιαιτερότητα της τοπογραφίας της μυϊκής ατροφίας, την παρουσία ή απουσία ψευδουπερτροφίας και συστολών τενόντων και άλλα σημεία.
Οι περισσότερες μυϊκές δυστροφίες είναι καλά μελετημένες κλινικά Λεπτομερής περιγραφήπου έγινε στα τέλη του περασμένου αιώνα. Όμως, παρά τον σχεδόν έναν αιώνα ιστορίας της μελέτης της μυοδυστροφίας, τα ζητήματα της παθογένειας και της αντιμετώπισής τους παραμένουν άλυτα μέχρι σήμερα. Μεγάλες ελπίδες εναποτίθενται στη μοριακή γενετική, με τη βοήθεια της οποίας έχει ήδη προσδιοριστεί η θέση των γονιδίων πολλών νοσολογικών μορφών.
Η διάγνωση των μυϊκών δυστροφιών συχνά παρουσιάζει μεγάλες δυσκολίες. Υπάρχει μεγάλη μεταβλητότητα στις κλινικές εκδηλώσεις και ένας μικρός αριθμός μελών της οικογένειας καθιστά δύσκολο τον προσδιορισμό του είδους της κληρονομικότητας.
Ένα χαρακτηριστικό κινητικό ελάττωμα σε ασθενείς με μυϊκές δυστροφίες είναι το βάδισμα «πάπιας»: ο ασθενής περπατά κουνώντας προς τη μία πλευρά. Συνδέεται κυρίως με την αδυναμία των γλουτιαίων μυών, κυρίως των μεσαίων και μικρών, που στερεώνουν τη λεκάνη σε σχέση με το μηριαίο οστό. Ως αποτέλεσμα, η νόσος προκαλεί κλίση της λεκάνης προς το μη υποστηρικτικό πόδι (φαινόμενο Trendelenburg) και αντισταθμιστική κλίση του κορμού προς την αντίθετη κατεύθυνση (φαινόμενο Duchenne). Όταν περπατάτε, η πλευρά της πλαγιάς αλλάζει συνεχώς. Αυτές οι αλλαγές μπορούν επίσης να ελεγχθούν στο τεστ Trendelenburg ζητώντας από τον ασθενή να σηκώσει το ένα πόδι, λυγίζοντας το σε ορθή γωνία στις αρθρώσεις του γόνατος και του ισχίου: η λεκάνη στο πλάι του ανυψωμένου ποδιού πέφτει (και δεν σηκώνεται κανονικά). λόγω αδυναμίας του μέσου γλουτιαίου μυός του στηρικτικού ποδιού.
ανερχόμενος από οριζόντια θέση, ένας ασθενής με σοβαρή μυϊκή αδυναμία των εγγύς μυών σχεδόν κυλάει στο στομάχι του, στη συνέχεια, ακουμπώντας τα χέρια του στο πάτωμα, ανεβαίνει στα τέσσερα και μετά, ακουμπώντας τα χέρια του στις κνήμες και μετά στους γοφούς, σταδιακά ισιώνει . Αυτό το φαινόμενο «διαλέγοντας μόνος σου» ονομάζεται ελιγμός Gowers. Συχνά σχετίζεται με αδυναμία των μυών του μεγίστου γλουτιαίου.
Μυοδυστροφία Duchenne.Η ψευδοϋπερτροφική μυϊκή δυστροφία Duchenne εμφανίζεται συχνότερα από όλες τις άλλες παθήσεις του μυϊκού συστήματος (30 ανά 100.000 γεννήσεις ζώντων). Χαρακτηρίζεται από πρώιμη έναρξη και κακοήθη πορεία. Η κλασική εικόνα εκδηλώνεται με αλλαγή στο βάδισμα σε ένα παιδί ηλικίας 2-5 ετών, στην ηλικία των 8-10 ετών τα παιδιά περπατούν ήδη με δυσκολία, στην ηλικία των 14-15 ετών είναι συνήθως εντελώς ακινητοποιημένα. Τα παιδιά έχουν περισσότερα Νεαρή ηλικίατα αρχικά συμπτώματα εκδηλώνονται με καθυστέρηση στην κινητική ανάπτυξη: αρχίζουν να περπατούν αργότερα, δεν μπορούν να τρέξουν και να πηδήξουν. Οι ασθενείς πεθαίνουν στη 2η ή 3η δεκαετία της ζωής τους.
Ένα από τα πρώτα σημάδια της νόσου είναι η συμπίεση των μυών της γάμπας και η σταδιακή αύξηση του όγκου τους λόγω ψευδουπερτροφίας. Η ατροφία των μυών του μηρού, της πυελικής ζώνης συχνά καλύπτεται από καλά ανεπτυγμένο υποδόριο λιπώδη ιστό. Σταδιακά, η διαδικασία παίρνει μια ανοδική κατεύθυνση και εξαπλώνεται πέρα από την ωμική ζώνη, τους μύες της πλάτης και στη συνέχεια στα εγγύς μέρη των χεριών.
ΣΕ τερματικό στάδιοη μυϊκή αδυναμία μπορεί να εξαπλωθεί στους μύες του προσώπου, του φάρυγγα, των αναπνευστικών μυών.
Σε προχωρημένο στάδιο της νόσου, υπάρχουν χαρακτηριστικά συμπτώματασαν "πάπια βόλτα"? τονισμένη οσφυϊκή λόρδωση, πτερυγοειδείς ωμοπλάτες, ένα σύμπτωμα της «χαλαρής ζώνης ώμου». Οι πρώιμες συσπάσεις των μυών και οι συσπάσεις των τενόντων είναι χαρακτηριστικές, ειδικά για τους αχίλλειους τένοντες. Τα αντανακλαστικά του γόνατος πέφτουν νωρίς και μετά τα αντανακλαστικά από τα άνω άκρα.
Η ψευδουπερτροφία μπορεί να αναπτυχθεί όχι μόνο στο γαστροκνήμιο, αλλά και στους γλουτιαίους, δελτοειδή, κοιλιακούς και μύες της γλώσσας. Πολύ συχνά ο καρδιακός μυς υποφέρει από τον τύπο της μυοκαρδιοπάθειας. Αποκαλύπτονται παραβιάσεις του ρυθμού της καρδιακής δραστηριότητας, επέκταση των ορίων της καρδιάς, κώφωση των τόνων, αλλαγές στο ΗΚΓ. Η οξεία καρδιακή ανεπάρκεια είναι η μεγαλύτερη Κοινή αιτίαθανάτους στη μυοδυστροφία Duchenne. Στην αυτοψία διαπιστώνεται ίνωση και λιπώδης διήθηση του καρδιακού μυός.
Συχνά υπάρχει παραβίαση της κινητικότητας του γαστρεντερικού σωλήνα.
Κοινό σύμπτωμαείναι μείωση της νοημοσύνης. Ενδιαφέρον παρουσιάζει το γεγονός ότι σε κάποιες οικογένειες η ολιγοφρένεια εκφράζεται έντονα, σε άλλες σχετικά μέτρια. Αλλαγή σε υψηλότερα νοητικές λειτουργίεςσυνήθως δεν εξελίσσεται και δεν συσχετίζεται με τη σοβαρότητα του μυϊκού ελαττώματος. Δεν μπορεί να εξηγηθεί μόνο από την παιδαγωγική παραμέληση των άρρωστων παιδιών που αποκλείονται πρόωρα από τις παιδικές ομάδες, δεν φοιτούν νηπιαγωγείοκαι το σχολείο λόγω κινητικών αναπηριών. Η αξονική τομογραφία και η μαγνητική τομογραφία συχνά αποκαλύπτουν εγκεφαλική ατροφία, που πιθανώς σχετίζεται με διαταραχή της προγεννητικής ανάπτυξης του εγκεφάλου.
Συχνά, τα παιδιά αναπτύσσουν λιπογεννητικό σύνδρομο, μερικές φορές άλλα σημάδια ενδοκρινικής ανεπάρκειας. Συχνά εντοπίζουν αλλαγές στο σκελετικό σύστημα: παραμόρφωση των ποδιών, στήθος, σπονδυλική στήλη, διάχυτη οστεοπόρωση.
Διακριτικό χαρακτηριστικόΗ μορφή Duchenne είναι υψηλός βαθμόςυπερενζυμαιμία ήδη πρώιμα στάδιαανάπτυξη της διαδικασίας. Έτσι, το επίπεδο ενός ενζύμου που είναι ειδικό για τον μυϊκό ιστό - η φωσφοκινάση της κρεατινίνης - στον ορό του αίματος μπορεί να υπερβεί δεκάδες και ακόμη και εκατοντάδες φορές κανονική απόδοση. Μια απότομη (10-100 φορές) αύξηση της φωσφοκινάσης κρεατινίνης (CPK) στη νευρομυϊκή παθολογία θα πρέπει να προκαλέσει συζήτηση, πρώτα από όλα τις ακόλουθες ασθένειες: Νόσος Duchenne, νόσος Becker, πολιομυοσίτιδα και δερματομυοσίτιδα, παροξυσμική μυοσφαιρινουρία, περιφερική μυοδυστροφία. Μόνο στα προχωρημένα στάδια της νόσου, ο βαθμός υπερενζυμαιμίας μειώνεται σταδιακά. Υπάρχουν αναφορές για αύξηση της CPK στο στάδιο της ενδομήτριας ανάπτυξης.
Η μυϊκή δυστροφία Duchenne μεταδίδεται με υπολειπόμενο τρόπο που συνδέεται με Χ. Το γονίδιο βρίσκεται στο κοντό βραχίονα του χρωμοσώματος Χ. Η συχνότητα της γονιδιακής μετάλλαξης είναι αρκετά υψηλή (30%), γεγονός που εξηγεί ένας μεγάλος αριθμός απόσποραδικές περιπτώσεις.
Μια μετάλλαξη (τις περισσότερες φορές μια διαγραφή) οδηγεί σε σεξουαλική ή σχεδόν πλήρη απουσία ενός γονιδιακού προϊόντος - μιας δομικής πρωτεΐνης δυστροφικής. Ο φυσιολογικός ρόλος του δυστροφικού δεν έχει πλήρως τεκμηριωθεί. Βρίσκεται σε υψηλές συγκεντρώσεις στο σαρκόλημμα, παίζοντας προφανώς ρόλο στη διατήρηση της ακεραιότητας αυτής της μεμβράνης. Η απουσία δυστροφικής προκαλεί δομικές αλλαγές στο σαρκόλημμα, το οποίο με τη σειρά του οδηγεί σε απώλεια ενδοκυτταρικών συστατικών και αυξημένη πρόσληψη ασβεστίου, που τελικά οδηγεί στο θάνατο των μυοϊνιδίων. Πιστεύεται ότι η ανεπάρκεια δυστροφικής στις συναπτικές ζώνες των νευρώνων του φλοιού είναι η αιτία της νοητικής καθυστέρησης.
Για την ιατρική γενετική συμβουλευτική, η καθιέρωση ετερόζυγου φορείου είναι πολύ σημαντική. Με τη μυοδυστροφία Duchenne σε ετεροζυγώτες, σε περίπου 70% των περιπτώσεων, ανιχνεύονται υποκλινικά και μερικές φορές εμφανή σημεία μυϊκής παθολογίας - κάποια συμπίεση και ακόμη και αύξηση στους μύες της γάμπας, ταχεία μυϊκή κόπωση κατά τη σωματική άσκηση, αλλαγές στο ΗΜΓ και παθομορφολογική εξέταση των μυών δείγματα βιοψίας. Τις περισσότερες φορές, οι ετερόζυγοι φορείς παρουσιάζουν αύξηση της δραστηριότητας της φωσφοκινάσης της κρεατινίνης.
Με την παρουσία κλινικής εικόνας της μυοδυστροφίας Duchenne στις γυναίκες, θα πρέπει πρώτα να αποκλειστεί η πιθανότητα ανωμαλίας στο χρωμόσωμα Χ - σύνδρομο Shereshevsky-Turner (XO), σύνδρομο Morris (XY) ή μωσαϊκό σε αυτά τα σύνδρομα.
Η μυϊκή δυστροφία Duchenne, η οποία αρχίζει να αναπτύσσεται ακόμη και στην προγεννητική περίοδο, είναι ουσιαστικά μια συγγενής μυοπάθεια και μπορεί να διαγνωστεί λίγο μετά τη γέννηση με τη διενέργεια βιοψίας μυών και τον προσδιορισμό της δραστηριότητας της CPK.
Μυοδυστροφία Becker.Μαζί με βαριά κακοήθης μορφήΥπάρχει μια καλοήθης μορφή μυϊκής δυστροφίας Duchenne που συνδέεται με Χ - η νόσος του Becker. Όσον αφορά τα κλινικά συμπτώματα, μοιάζει πολύ με τη μορφή Duchenne, αλλά, κατά κανόνα, αρχίζει αργότερα - σε ηλικία 10-15 ετών, ρέει απαλά, οι ασθενείς παραμένουν σε θέση να εργαστούν για μεγάλο χρονικό διάστημα, στην ηλικία των 20 ετών -30 χρόνια και μετά μπορούν ακόμα να περπατήσουν. Η γονιμότητα δεν μειώνεται, επομένως η ασθένεια εντοπίζεται μερικές φορές σε πολλές γενιές της οικογένειας: ένας άρρωστος άνδρας μεταδίδει την ασθένεια στον εγγονό του μέσω της κόρης του («φαινόμενο του παππού»). Τα αρχικά συμπτώματα, όπως και στη νόσο Duchenne, εκδηλώνονται με αδυναμία στους μύες της πυελικής ζώνης και στη συνέχεια στα εγγύς κάτω άκρα. Οι ασθενείς αλλάζουν το βάδισμά τους, αντιμετωπίζουν δυσκολία όταν ανεβαίνουν σκάλες, όταν σηκώνονται από ένα χαμηλό κάθισμα. Χαρακτηρίζεται από ψευδουπερτροφία των μυών της γάμπας. Η συστολή των τενόντων της πτέρνας (Αχίλλειος) είναι λιγότερο έντονη από ό,τι στη νόσο Duchenne.
Με αυτή τη μορφή, δεν υπάρχουν διανοητικές βλάβες, η μυοκαρδιοπάθεια απουσιάζει ή εκφράζεται ελαφρώς.
Όπως και με άλλες μυοδυστροφίες που συνδέονται με Χ, η μορφή του Becker αυξάνει σημαντικά τη δραστηριότητα της CPK, αν και σε μικρότερο βαθμό από ό,τι στο Duchenne, που δεν υπερβαίνει τις 5000 μονάδες. Το γονίδιο για τη νόσο του Becker, όπως και η νόσος του Duchenne, εντοπίζεται στο κοντό βραχίονα του χρωμοσώματος Χ. είναι πιθανό και οι δύο τόποι να σχετίζονται στενά ή να είναι αλληλόμορφοι. Σε αντίθεση με τη νόσο του Duchenne, στην οποία πρακτικά δεν υπάρχει δυστροφία, η ανώμαλη δυστροφία συντίθεται στη νόσο του Becker. Διαφορές εντοπίζονται επίσης στη βιοψία μυών. Με τη μυϊκή δυστροφία του Becker, οι μυϊκές ίνες συνήθως δεν είναι στρογγυλές, οι υαλώδεις ίνες, χαρακτηριστικές της μυϊκής δυστροφίας του Duchenne, είναι εξαιρετικά σπάνιες.
Μυοδυστροφία Landouzy-Dejerine (μυοδυστροφία προσώπου-ώμου).Η νόσος μεταδίδεται με αυτοσωμικό κυρίαρχο τρόπο με υψηλή διεισδυτικότητα αλλά κάπως μεταβλητή εκφραστικότητα. Εμφανίζεται πολύ λιγότερο συχνά από τη μυοδυστροφία Duchenne (0,4 ανά 100 χιλιάδες πληθυσμού). Πιστεύεται ότι το γονίδιο αυτής της ασθένειας εντοπίζεται στο 4ο χρωμόσωμα. Οι γυναίκες αρρωσταίνουν πιο συχνά από τους άνδρες (3:1), σωματική υπερφόρτωση, εντατικά αθλήματα, καθώς και παράλογα φυσιοθεραπείαμπορεί να συμβάλει σε πιο σοβαρή πορεία της νόσου.
Μυοδυστροφία Landouzy-Dejerine - σχετικά ευνοϊκή τρέχουσα μορφήμυϊκή παθολογία. Ξεκινά σε ηλικία περίπου 20 ετών, μερικές φορές αργότερα. Ωστόσο, σε οικογενειακές υποθέσειςασθένειες, όταν μπορείτε να παρακολουθήσετε τη δυναμική των νεότερων μελών της οικογένειας, είναι δυνατό να εντοπιστεί κάποια αδυναμία των μυών, για παράδειγμα, οι μύες του προσώπου και σε μικρότερη ηλικία.
Η μυϊκή αδυναμία και η ατροφία εμφανίζονται αρχικά στους μύες του προσώπου ή της ωμικής ζώνης. Σταδιακά, αυτές οι διαταραχές εξαπλώθηκαν στους μύες των εγγύς βραχιόνων και στη συνέχεια στους μύες των εγγύς βραχιόνων κάτω άκρα. Στις περισσότερες περιπτώσεις, προσβάλλονται πρώτα οι μύες της πρόσθιας επιφάνειας των ποδιών (με την ανάπτυξη ενός κρεμαμένου ποδιού), στη συνέχεια οι μύες των εγγύς ποδιών. Στο ύψος της νόσου προσβάλλονται σοβαρά οι κυκλικοί μύες του ματιού και του στόματος, ο μείζονος θωρακικός, ο πρόσθιος οδοντωτός και το κάτω τμήμα του τραπεζοειδούς μυός, latissimus dorsiπλάτη, δικέφαλος, τρικέφαλος βραχιόνιος. χαρακτηριστικό γνώρισμα εμφάνισηασθενείς: τυπικό πρόσωπομυοπαθής με «εγκάρσιο χαμόγελο» («La Gioconda’s smile»), προεξοχή άνω χείλος(«ταπίρ χείλη»), έντονες πτερυγοειδείς ωμοπλάτες, μια ιδιόμορφη παραμόρφωση του θώρακα με την επιπέδωσή του στην προσθιοοπίσθια κατεύθυνση και την περιστροφή προς τα μέσα αρθρώσεις ώμων. Συχνά υπάρχει ασυμμετρία της βλάβης, ακόμη και μέσα στον ίδιο μυ (για παράδειγμα, κυκλικός μυςστόμα). Μπορεί να παρατηρηθεί ψευδουπερτροφία του γαστροκνήμιου, των δελτοειδών μυών και μερικές φορές των μυών του προσώπου. Οι συσπάσεις και οι συσπάσεις εκφράζονται μέτρια. τενοντιακά αντανακλαστικά πολύς καιρόςδιατηρούνται, αλλά μερικές φορές μειώνονται σε πρώιμο στάδιο.
Τα σημάδια βλάβης στον καρδιακό μυ είναι σπάνια. Η ενζυμική δραστηριότητα του ορού είναι ελαφρώς αυξημένη και μπορεί να είναι φυσιολογική. Η διάνοια δεν υποφέρει. Το προσδόκιμο ζωής στις περισσότερες περιπτώσεις δεν μειώνεται. Ενδιαφέρον παρουσιάζει το γεγονός ότι το ΗΜΓ στη μυοδυστροφία Landouzy-Dejerine συχνά δεν είναι αρκετά τυπικό για το μυϊκό επίπεδο της βλάβης. Σε ορισμένους ασθενείς (μέλη της ίδιας οικογένειας), μπορεί να παρατηρηθεί μείωση του εύρους των βιοδυναμικών, ένας τύπος παρεμβολής της καμπύλης, σε άλλους, αντίθετα, μείωση της συχνότητας και της υπερσύγχρονης δραστηριότητας, μερικές φορές με τυπικό πικετό ρυθμός φράχτη. Θα πρέπει να θυμόμαστε για την παραλλαγή της σπονδυλικής στήλης, η οποία μιμείται τη νόσο Landouzy-Dejerine.
Μυοδυστροφία Erb-Roth (μυοδυστροφία άκρου-ζώνης).Μεταδιδόμενα με αυτοσωμικό υπολειπόμενο τρόπο, προσβάλλονται και τα δύο φύλα εξίσου. Η εμφάνιση της νόσου στις περισσότερες περιπτώσεις αναφέρεται στα μέσα της 2ης δεκαετίας της ζωής (14-16 ετών), ωστόσο περιγράφεται ως πρώιμη, ψευδο-Duchenne μορφή, όταν τα πρώτα συμπτώματα εμφανίζονται πριν από την ηλικία των 10 ετών και η νόσος είναι σοβαρή και όψιμη παραλλαγή με έναρξη μετά από 30 χρόνια.
Η πορεία της νόσου μπορεί να είναι γρήγορη ή πιο αργή, κατά μέσο όρο, η πλήρης αναπηρία εμφανίζεται μέσα σε 15-20 χρόνια από την εμφάνιση των πρώτων συμπτωμάτων. Η μυοδυστροφία ξεκινά είτε με βλάβη στους μύες της πυελικής ζώνης και των εγγύς ποδιών (μορφή Leiden-Mobius), είτε από την ωμική ζώνη (μορφή Erb). Σε ορισμένες περιπτώσεις, ο ώμος και η πυελική ζώνη επηρεάζονται ταυτόχρονα. Οι μύες της πλάτης και της κοιλιάς υποφέρουν αρκετά σημαντικά. Οι ασθενείς έχουν χαρακτηριστικό βάδισμα «πάπιας», είναι δύσκολο να σηκωθούν από ξαπλωμένη και καθιστή θέση, τόνισε οσφυϊκή λόρδωση. Οι μύες του προσώπου δεν επηρεάζονται στις περισσότερες περιπτώσεις. Για αυτή τη μορφή, οι συσπάσεις και η ψευδουπερτροφία δεν είναι χαρακτηριστικές. Μπορεί να εμφανιστούν τελικές ατροφίες και συστολές τενόντων. Η νοημοσύνη συνήθως διατηρείται. Ο καρδιακός μυς είναι ως επί το πλείστον ανεπηρέαστος. Το επίπεδο των ενζύμων στον ορό του αίματος, κατά κανόνα, είναι αυξημένο, αλλά όχι τόσο απότομα όσο στην Χ-συνδεδεμένη μυοδυστροφία. Υπάρχουν ενδείξεις ότι στους άνδρες ασθενείς το επίπεδο της CPK είναι υψηλότερο από ότι στις γυναίκες ασθενείς. Υπάρχει σημαντική διαφορά στην έκφραση του μεταλλαγμένου γονιδίου σε διαφορετικά μέληοικογένεια - μαζί με σοβαρή κλινική εικόναμπορεί να υπάρχουν σχετικά ήπια και ακόμη και διαγραμμένα κλινικά συμπτώματα. Ο θάνατος επέρχεται συνήθως από πνευμονικές επιπλοκές.
Δεδομένου ότι η κλινική μυοδυστροφίας άκρου-ζώνης είναι ιδιαίτερα πρόθυμη να μιμηθεί νευρομυϊκές παθήσειςδιαφορετικής φύσης, είναι απαραίτητο, ειδικά σε σποραδικές περιπτώσεις και με όψιμη έναρξη της νόσου, να γίνει ενδελεχής κλινική εξέταση για να αποκλειστούν νωτιαία αμυοτροφία, πολυμυοσίτιδα, μεταβολικές, ενδοκρινικές, τοξικές, φαρμακευτικές, καρκινωματώδεις μυοπάθειες. Στο παρελθόν, υπήρξε σαφής υπερδιάγνωση αυτής της μορφής μυϊκής δυστροφίας.
Θεραπεία μυϊκών δυστροφιών.Οι θεραπευτικές επιλογές για τις μυϊκές δυστροφίες είναι πολύ περιορισμένες. Αιτιολογική και παθογενετική θεραπείαπρακτικά ανύπαρκτο. Η συμπτωματική θεραπεία στοχεύει κυρίως στην πρόληψη της ανάπτυξης συσπάσεων, στη διατήρηση της υπάρχουσας μυϊκής δύναμης και, πιθανώς, σε κάποια μείωση του ρυθμού ατροφίας. Το κύριο καθήκον είναι να μεγιστοποιηθεί η περίοδος κατά την οποία ο ασθενής μπορεί να κινηθεί ανεξάρτητα, αφού μέσα ξαπλωμένη θέσηοι συσπάσεις, η σκολίωση, οι αναπνευστικές διαταραχές αυξάνονται ραγδαία. Ιατρικό συγκρότημαθα πρέπει να περιλαμβάνει θεραπευτικές ασκήσεις, μασάζ, ορθοπεδικά μέτρα, φαρμακευτική θεραπεία.
Η θεραπευτική γυμναστική αποτελείται από παθητικές και ενεργητικές κινήσεις που εκτελούνται σε όλες τις αρθρώσεις σε διάφορες στάσεις: όρθια, καθιστή, ξαπλωμένη, με διάφορες θέσεις των άκρων. Οι ενεργές κινήσεις εκτελούνται κατά προτίμηση σε ισομετρική λειτουργία. Η γυμναστική πρέπει να γίνεται τακτικά πολλές φορές την ημέρα. Ταυτόχρονα, θα πρέπει να προειδοποιηθεί κανείς για υπερβολικές ασκήσεις, ειδικά αυτές που συνοδεύονται από υπερβολική διάταση των μυών. Σημασια(ιδιαίτερα μετά την ακινητοποίηση του ασθενούς) κάντε ασκήσεις αναπνοής.
Ορθοπεδικά μέτρα συντηρητικού (ειδικά ελαστικά) και λειτουργικής φύσης (Αχιλλειοτομή, διασταύρωση μυς της γάμπας), που στοχεύουν στη διόρθωση των συσπάσεων και των αναδυόμενων παθολογικών στάσεων των άκρων, στοχεύουν επίσης στη διατήρηση της δυνατότητας ανεξάρτητης κίνησης. Σε κάθε περίπτωση, είναι απαραίτητο να σταθμιστούν μεμονωμένα τα αναμενόμενα οφέλη και πιθανή βλάβηαπό χειρουργική επέμβαση. Θα πρέπει να ληφθεί υπόψη ότι συχνά (ιδιαίτερα, με σοβαρή υπερλόρδωση και αδυναμία του τετρακέφαλου μηριαίου μυός), η ισημερινή θέση των ποδιών είναι αντισταθμιστικής σημασίας και μετά, για παράδειγμα, αχιλοτομή, ο ασθενής μπορεί να ακινητοποιηθεί πλήρως. Με την ανάπτυξη συσπάσεων, συνιστάται να τεντώνετε προσεκτικά τους μύες έως και 20-30 φορές την ημέρα, ακολουθούμενο από νάρθηκα κατά τη διάρκεια του ύπνου.
Ιατρική θεραπείαπεριλαμβάνει το διορισμό μεταβολικών φαρμάκων που στοχεύουν στην κάλυψη της ανεπάρκειας ενέργειας και πρωτεΐνης, αλλά η αποτελεσματικότητά τους είναι πολύ αμφίβολη. Χρησιμοποιούνται ανταγωνιστές ασβεστίου (λόγω ελαττώματος στις κυτταρικές μεμβράνες που εντοπίστηκε στη νόσο Duchenne, που οδηγεί σε αυξημένη πρόσληψη ασβεστίου στο κύτταρο), ανοσοτροποποιητές, ενώσεις που περιέχουν φώσφορο (ATP, φωσφαδένη), βιταμίνη Ε (100 mg από το στόμα 3 φορές την ημέρα). ημέρα). Έχει αποδειχθεί ότι η χρήση πρεδνιζολόνης (0,75 mg/kg ημερησίως) στη νόσο Duchenne μπορεί να αυξήσει δραματικά τη μυϊκή δύναμη, αλλά αυτή η επίδραση παραμένει για όχι περισσότερο από ένα χρόνο και γενικά δεν επηρεάζει την έκβαση της νόσου. Λόγω σοβαρών παρενέργειες, που προκύπτει vri μακροχρόνια χρήσηφάρμακο, η χρήση του είναι ακατάλληλη. Οι εκτιμήσεις για την επίδραση των αναβολικών στεροειδών είναι αντιφατικές και η συνταγογράφηση τους συνδέεται συχνά με αδικαιολόγητο κίνδυνο. Κατά την αξιολόγηση της επίδρασης ορισμένων φαρμάκων στο Duchenne, θα πρέπει να ληφθεί υπόψη ότι με μέτρια βαρύτητα της νόσου σε ασθενείς ηλικίας 3-6 ετών, μπορεί να υπάρξει σχετική σταθεροποίηση της κατάστασης που σχετίζεται με ηλικιακή ανάπτυξημυϊκό σύστημα, η απόκτηση κινητικών δεξιοτήτων, που μπορούν σε κάποιο βαθμό να αντισταθμίσουν προσωρινά τη συνεχώς συνεχιζόμενη δυστροφική διαδικασία.
Καθορισμένη αξίαέχει διόρθωση της διατροφής του ασθενούς, συνιστάται δίαιτα υψηλή σε πρωτεΐνες και χαμηλή σε λιπαρά και μειωμένη σε θερμίδες με βέλτιστη περιεκτικότητα σε βιταμίνες και μικροστοιχεία. Σημαντικό ρόλο παίζει η ψυχολογική υποστήριξη του ασθενούς, η συνέχιση της εκπαίδευσης και ο σωστός επαγγελματικός προσανατολισμός.
Οι νευρομυϊκές παθήσεις (NMD) είναι η πολυπληθέστερη ομάδα κληρονομικών ασθενειών, οι οποίες βασίζονται σε γενετικά καθορισμένη βλάβη στα πρόσθια κέρατα του νωτιαίου μυελού, στα περιφερικά νεύρα και στους σκελετικούς μύες.
Οι νευρομυϊκές παθήσεις περιλαμβάνουν:
1) προοδευτικές μυϊκές δυστροφίες (πρωτοπαθείς μυοπάθειες).
2) σπονδυλικές και νευρικές αμυοτροφίες (δευτερογενείς μυοπάθειες).
3) συγγενείς μη προοδευτικές μυοπάθειες.
4) νευρομυϊκές παθήσεις με μυοτονικό σύνδρομο.
5) παροξυσμική μυοπληγία.
6) μυασθένεια gravis.
15.2. Προοδευτικές μυϊκές δυστροφίες (πρωτοπαθείς μυοπάθειες)
Προοδευτικές μυϊκές δυστροφίες (PMD),ή πρωτοπαθείς μυοπάθειες, χαρακτηρίζονται από εκφυλιστικές αλλαγές στον μυϊκό ιστό.
Παθολογικές αλλαγέςΤο PMD χαρακτηρίζεται από λέπτυνση των μυών, αντικαθιστώντας τους με λιπώδη και συνδετικό ιστό. Οι εστίες της εστιακής νέκρωσης αποκαλύπτονται στο σαρκόπλασμα, οι πυρήνες των μυϊκών ινών είναι διατεταγμένοι σε αλυσίδες, οι μυϊκές ίνες χάνουν την εγκάρσια ραβδώσεις τους.
Τα θέματα παθογένειας παραμένουν άλυτα μέχρι σήμερα. Η μυοπάθεια βασίζεται σε ένα ελάττωμα στη μεμβράνη των μυϊκών κυττάρων. Μεγάλες ελπίδες εναποτίθενται στη μοριακή γενετική.
Οι διάφορες μορφές μυοπάθειας διαφέρουν ως προς τον τύπο της κληρονομικότητας, τον χρόνο έναρξης της διαδικασίας, τη φύση και την ταχύτητα της πορείας της και την τοπογραφία της μυϊκής ατροφίας.
Οι μυοπάθειες χαρακτηρίζονται κλινικά από μυϊκή αδυναμία και ατροφία. Υπάρχουν διάφορες μορφές PMD.
15.2.1. Μυοδυστροφία Duchenne (ψευδουπερτροφική μορφή PMD)
Εμφανίζεται πιο συχνά από όλα τα PMD (30:100.000). Αυτή η μορφή χαρακτηρίζεται από πρώιμη έναρξη (2-5 ετών) και κακοήθη πορεία, κυρίως τα αγόρια νοσούν. Η μυοπάθεια Duchenne κληρονομείται με υπολειπόμενο τρόπο που συνδέεται με Χ. Το παθολογικό γονίδιο εντοπίζεται στο κοντό βραχίονα του χρωμοσώματος (Χ, ή χρωμόσωμα 21).
Η μετάλλαξη του γονιδίου είναι αρκετά υψηλή, γεγονός που εξηγεί τη σημαντική συχνότητα των σποραδικών περιπτώσεων. Η μετάλλαξη (τις περισσότερες φορές διαγραφή) ενός γονιδίου οδηγεί στην απουσία δυστροφίνης στη μεμβράνη μυϊκά κύτταρα, που οδηγεί σε δομικές αλλαγές στο σαρκόλημμα. Αυτό προάγει την απελευθέρωση ασβεστίου και οδηγεί στο θάνατο των μυοϊνιδίων.
Ένα από τα πρώτα σημάδια της νόσου είναι η συμπίεση των μυών της γάμπας και η σταδιακή αύξηση του όγκου τους λόγω ψευδουπερτροφίας. Η διαδικασία είναι ανοδική. Το προχωρημένο στάδιο της νόσου χαρακτηρίζεται από βάδισμα «πάπιας», ο ασθενής περπατά, κουνιέται από πλευρά σε πλευρά, που οφείλεται κυρίως σε αδυναμία των γλουτιαίων μυών.
Ως αποτέλεσμα, παρατηρείται κλίση της λεκάνης προς το μη υποστηρικτικό πόδι (φαινόμενο Trendelenburg) και αντισταθμιστική κλίση του κορμού προς την αντίθετη κατεύθυνση (φαινόμενο Duchenne). Όταν περπατάτε, η πλευρά της πλαγιάς αλλάζει συνεχώς. Αυτό μπορεί να ελεγχθεί στη θέση Trendelenburg ζητώντας από τον ασθενή να σηκώσει το ένα πόδι, λυγίζοντας το σε ορθή γωνία στην άρθρωση του γόνατος και του ισχίου: η λεκάνη στο πλάι του σηκωμένου ποδιού πέφτει (και δεν σηκώνεται κανονικά) λόγω αδυναμία του μέσου γλουτιαίου μυός του στηρικτικού ποδιού.
Με μυοπάθεια Duchenne, έντονη λόρδωση, πτερυγοειδείς ωμοπλάτες, τυπικές μυϊκές συσπάσεις και τραντάγματα του γόνατος συχνά πέφτουν νωρίς. Συχνά είναι δυνατό να εντοπιστούν αλλαγές στο σκελετικό σύστημα (παραμόρφωση ποδιών, θώρακα, σπονδυλικής στήλης, διάχυτη οστεοπόρωση). Μπορεί να υπάρξει μείωση της νοημοσύνης και διάφορες ενδοκρινικές διαταραχές (λιπογεννητικό σύνδρομο, σύνδρομο Itsenko-Cushing). Στην ηλικία των 14-15 ετών, οι ασθενείς είναι συνήθως ήδη εντελώς ακινητοποιημένοι· στο τελικό στάδιο, η αδυναμία μπορεί να εξαπλωθεί στους μύες του προσώπου, του φάρυγγα και του διαφράγματος. Πεθαίνουν συχνότερα την 3η δεκαετία της ζωής τους από μυοκαρδιοπάθεια ή από προσθήκη παρεπόμενων λοιμώξεων.
Ένα χαρακτηριστικό γνώρισμα της μυοπάθειας Duchenne είναι η απότομη αύξηση ενός συγκεκριμένου μυϊκού ενζύμου - κρεατινοφωσφοκινάση (CPK) κατά δεκάδες και εκατοντάδες φορές, καθώς και αύξηση της μυοσφαιρίνης κατά 6-8 φορές.
Για την ιατρική γενετική συμβουλευτική, είναι σημαντικό να καθιερωθεί ετερόζυγος φορέας. Στο 70% των ετεροζυγωτών προσδιορίζονται υποκλινικά και κλινικά σημεία μυϊκής παθολογίας: πάχυνση και διεύρυνση των μυών της γάμπας, ταχεία μυϊκή κόπωση κατά τη σωματική άσκηση, αλλαγές στις μυϊκές βιοψίες και βιοδυναμίες σύμφωνα με δεδομένα ΗΜΓ.
Σελίδα 44 από 44
Εμπλέκονται οι σκελετικοί μύες παθολογική διαδικασίαμε ποικιλία εκφυλιστικών, μεταβολικών και φλεγμονώδεις ασθένειες. Στις περισσότερες περιπτώσεις, αυτό οδηγεί σε εκφυλισμό. μυϊκές ίνες, και σε χρόνιες μορφές - την αντικατάστασή τους με συνδετικό ιστό και λίπος. Οι εγγύς μυϊκές ομάδες καταστρέφονται περισσότερο από τις άπω, καθώς και τα κάτω άκρα σε σχέση με τα άνω. Ένα άρρωστο παιδί διακρίνεται από το λεγόμενο βάδισμα της πάπιας, δεν μπορεί να τρέξει, να ανέβει σκάλες και να σηκωθεί αν είναι σε καθιστή θέση. Τα τενοντιακά του αντανακλαστικά είναι καταθλιπτικά, ο βαθμός εξαφάνισής τους είναι ανάλογος με τον βαθμό αποδυνάμωσης της μυϊκής δύναμης. Η ευαισθησία δεν επηρεάζεται.
Οι διαγνωστικά πολύτιμες εργαστηριακές μέθοδοι περιλαμβάνουν τον προσδιορισμό της δραστικότητας των ενζύμων, ιδιαίτερα της κρεατινοφωσφοκινάσης, στον ορό. Αυτό το ένζυμο, το οποίο καταλύει την αντίδραση: φωσφοκρεατίνη + ADP-κρεατίνη + ATP, υπάρχει κυρίως στα εγκεφαλικά κύτταρα και στους μυϊκούς ιστούς. Σε ορισμένες διάχυτες μυϊκές ασθένειες, ιδιαίτερα τη μυϊκή δυστροφία, οι υπερβολικές ποσότητες της διεισδύουν στον μεσοκυττάριο χώρο και στο αίμα. Σε ασθενείς, η δραστηριότητα της γαλακτικής αφυδρογονάσης του ορού και της γλουταμινοοξαλοξικής τρανσαμινάσης είναι συνήθως αυξημένη, αλλά η ευρεία κατανομή τους σε άλλους ιστούς, συμπεριλαμβανομένου του ήπατος, μειώνει την ειδικότητα της εξέτασης. Συνήθως απαιτείται βιοψία μυϊκού ιστού για να διευκρινιστεί η διάγνωση.
Φλεγμονώδεις μυϊκές παθήσεις. Η φλεγμονή του μυϊκού ιστού συνοδεύει ορισμένες λοιμώξεις, ιδιαίτερα την τριχίνωση, την τοξοπλάσμωση και αυτές που προκαλούνται από τον ιό Coxsackie. Συχνά αποτελεί συστατικό ασθενειών κολλαγόνου, συμπεριλαμβανομένης της δερματομυοσίτιδας, του ερυθηματώδους λύκου, της οζώδους περιαρτηρίτιδας και της ρευματοειδούς αρθρίτιδας.
Πολυμυοσίτιδα. Η διάχυτη μεμονωμένη μυϊκή φλεγμονή άγνωστης αιτιολογίας ονομάζεται πολυμυοσίτιδα. Χαρακτηρίζεται από ταχέως προοδευτική πορεία, αδυναμία και πόνο στις εγγύς μυϊκές ομάδες. Συχνά, οι μύες του λαιμού εμπλέκονται στη διαδικασία και ως εκ τούτου γίνεται δύσκολο για το παιδί να σηκώσει το κεφάλι του και να το κρατήσει σε αυτή τη θέση. Τα εργαστηριακά σημάδια μυϊκής φλεγμονής περιλαμβάνουν αύξηση του ESR και του αριθμού των λευκοκυττάρων. Ωστόσο, η απουσία τους δεν αποκλείει την πολυμυοσίτιδα. Τα επίπεδα των ενζύμων στον ορό είναι συνήθως αυξημένα. Στη μυϊκή βιοψία προσδιορίζεται ο εκφυλισμός και η μερική αναγέννηση των ινών και η διήθησή τους από τα λεμφοειδή κύτταρα. Είναι δύσκολο να διαφοροποιηθεί η πολυμυοσίτιδα από τη μυϊκή δυστροφία και τη δερματομυοσίτιδα. Μπορεί να αντιπροσωπεύει άτυπη μορφήδερματομυοσίτιδα, αν και το ιστολογικό πρότυπο σε αυτές τις δύο καταστάσεις είναι κάπως διαφορετικό: η δερματομυοσίτιδα χαρακτηρίζεται από αγγειίτιδα, η οποία συνήθως απουσιάζει στην πολυμυοσίτιδα. Η πρόγνωση για το τελευταίο είναι κάπως πιο ευνοϊκή. Η θεραπεία με κορτικοστεροειδή συνοδεύεται από αποτέλεσμα, αλλά όταν ακυρωθούν, μπορεί να εμφανιστεί υποτροπή.
Προοδευτική οστεοποιητική μυοσίτιδα. Η αιτιολογία αυτής της σπάνιας νόσου του συνδετικού ιστού και των μυών είναι άγνωστη. Αναφέρεται ότι τα αδέρφια, συμπεριλαμβανομένων των διδύμων, πάσχουν από αυτό και μεταδίδεται σε συγγενείς εξ αίματος απευθείας. Πιστεύεται ότι η νόσος κληρονομείται με αυτοσωμικό κυρίαρχο τρόπο. Τα αγόρια αρρωσταίνουν 2-3 φορές πιο συχνά από τα κορίτσια.
Παθολογικά σημείαεξαρτώνται από το στάδιο της νόσου. Στα αρχικά στάδια, τοπικό οίδημα και φλεγμονώδεις κυτταρικές διηθήσεις εντοπίζονται σε μύες και τένοντες. Αργότερα, οι περιοχές της φλεγμονής αντικαθίστανται από κοκκιώδη ιστό και τελικά σχηματίζονται περιοχές χόνδρου και οστικού ιστού στις βλάβες.
Σχεδόν το 75% των άρρωστων παιδιών έχουν γενετικές ανωμαλίεςανάπτυξη, συχνότερα υποανάπτυξη των δακτύλων και αγκύλωση των φαλαγγών των πρώτων δακτύλων και υπανάπτυξη των πρώτων δακτύλων, πολυδακτυλία, καμπυλότητα των δακτύλων, συνδακτυλία (πόδια), παραμόρφωση αυτιάκώφωση, έλλειψη δοντιών. Οι ίδιες συγγενείς δυσπλασίες μπορεί να υπάρχουν σε συγγενείς του ασθενούς που δεν έχουν αναπτύξει προοδευτική νόσο του συνδετικού ιστού και των μυών. Η ηλικία στην οποία μπορεί να ξεκινήσει η μυοσίτιδα ποικίλλει από τη γέννηση έως τη μεγαλύτερη παιδική ηλικία. Συνήθως, διακρίνονται τρία στάδια: 1) περιορισμένα, συχνά ζεστά και μαλακά στην αφή, παστώδεις διογκώσεις των μαλακών ιστών εμφανίζονται στα σημεία των μικροτοπικών τραυματισμών. 2) μετά από μερικές ημέρες, τα συμπτώματα της φλεγμονής εξαφανίζονται και η βλάβη σκληραίνει. 3) εμφανίζεται οστεοποίηση της πληγείσας περιοχής. Περιοδικά εμφανίζονται νέες βλάβες, κυρίως στον αυχένα και την πλάτη. πρωτογενές σύμπτωμαΤουρτικολίς μπορεί να γίνει εάν η διαδικασία έχει αναπτυχθεί στον στερνοκλειδομαστοειδή μυ. Τελικά, η οστεοποίηση επεκτείνεται σε πολλούς τένοντες και συνδέσμους. Εμφανίζεται αγκύλωση της σπονδυλικής στήλης και των αρθρώσεων των χεριών και των ποδιών (Εικ. 21-5). Η φλεγμονή μπορεί να εξαπλωθεί στις κροταφογναθικές αρθρώσεις, γεγονός που δυσκολεύει τις κινήσεις της μάσησης. Οι εκβολές των οστών μπορεί να προεξέχουν μέσα από το δέρμα. Στην εφηβεία, η ασθένεια συχνά οδηγεί σε πλήρη ακινητοποίηση και θάνατο λόγω αναπνευστική ανεπάρκειακαι διακοπή της αναπνοής, αν και υπάρχουν αναφορές για περιπτώσεις επιβίωσης. Με την οστεοποιητική μυοσίτιδα, υπάρχει υψηλός κίνδυνος ανάπτυξης οστεογενούς σαρκώματος.
Ρύζι. 21-5. Παιδί με προοδευτική οστεοειδίτιδα μυοσίτιδα (τυπική στάση με δυσκαμψία στον αυχένα και την πλάτη).
Μερικές φορές η παθολογική διαδικασία περιορίζεται στο σημείο ενός προηγούμενου τραυματισμού των μαλακών ιστών (miositis ossificans circumscripta). Εκτεταμένη ασβεστοποίηση του μυϊκού ιστού μπορεί επίσης να συμβεί σε χρόνια πολυμυοσίτιδα και δερματομυοσίτιδα.
Αποτελέσματα εργαστηριακές μεθόδουςοι μελέτες δεν έχουν διαγνωστική αξία.
Τα επίπεδα ασβεστίου, φωσφόρου, αλκαλικής φωσφατάσης στον ορό, καθώς και η δραστηριότητα της φωσφοκινάσης της κρεατίνης και άλλων ενζύμων παραμένουν φυσιολογικά. Ο οστικός ιστός στη βλάβη δεν διαφέρει στη δομή από τον κανόνα.
Υφιστάμενες Μέθοδοιοι θεραπείες δεν είναι ικανοποιητικές. Σε ορισμένες περιπτώσεις, έχει παρατηρηθεί επιβράδυνση στην ανάπτυξη της νόσου με τη χρήση ACTH και άλλων κορτικοστεροειδών. Ο ρόλος τους σε τελικό αποτέλεσμαη θεραπεία είναι αμφισβητήσιμη.
Ενδοκρινικές και μεταβολικές μυοπάθειες. Η μυοπάθεια στον υπερθυρεοειδισμό είναι μια αρκετά σπάνια επιπλοκή. Χαρακτηρίζεται από πτώση, αμφοτερόπλευρη πάρεση μύες του προσώπουκαι μύες των εγγύς άκρων. Ταυτόχρονα, ορισμένα συμπτώματα του υπερθυρεοειδισμού μπορεί να καλύπτονται από μυϊκή αδυναμία, αλλά ταχυκαρδία, αυξημένη εφίδρωση και αυξημένη θυρεοειδής αδένας. Τα τενοντιακά αντανακλαστικά, σε αντίθεση με πολλές άλλες μορφές μυοπάθειας, παραμένουν φυσιολογικά. Μετά τη διόρθωση του υπερθυρεοειδισμού, η μυϊκή αδυναμία σταδιακά εξαφανίζεται.
Μυοπάθεια στον υποθυρεοειδισμό. Ο υποθυρεοειδισμός στα βρέφη μπορεί να σχετίζεται με μυϊκή αδυναμία και υπόταση. Σε μεγαλύτερα παιδιά με μυξοίδημα, οι μυϊκές συσπάσεις και η χαλάρωση επιβραδύνονται, σε ορισμένες περιπτώσεις σημειώνεται μυϊκή υπερτροφία (σύνδρομο Debre-Semelen). Ο συνδυασμός σημείων όπως μυϊκή αδυναμία και υπερτροφία υποδηλώνει μυϊκή δυστροφία.
Μυοπάθεια κατά τη θεραπεία με κορτικοστεροειδή. Μπορεί να περιπλέξει τη νόσο του Itsenko-Cushing, αλλά πιο συχνά αναπτύσσεται στη θεραπεία μεγάλων δόσεων συνθετικών στεροειδών. Η αδυναμία είναι ιδιαίτερα αισθητή στους μύες της πυελικής ζώνης, η οποία εκδηλώνεται με βάδισμα με κωπηλασία (πάπια), δυσκολία στο ανέβασμα σκαλοπατιών και στην προσπάθεια να σηκωθείτε από καθιστή θέση. Το τράνταγμα στο γόνατο απουσιάζει. Μπορεί να εμφανιστεί αραίωση μυών. Οι μυοπαθητικές αλλαγές στον μυϊκό ιστό είναι συνήθως ασήμαντες ακόμη και με σοβαρή αδυναμία. Η μυϊκή δύναμη μετά την απόσυρση των κορτικοστεροειδών αποκαθίσταται αργά (μέσα σε αρκετούς μήνες).
Μυοπάθεια στον υπερπαραθυρεοειδισμό. Ο υπερπαραθυρεοειδισμός μπορεί να σχετίζεται με αδυναμία και υποαντανακλαστικότητα λόγω υπερκαλιαιμίας. Συνήθως εξαφανίζονται γρήγορα μετά την παραθυρεοειδεκτομή.
Η ανεπάρκεια καρνιτίνης (λιπιδική μυοπάθεια) συνοδεύει τη συσσώρευση μεγάλων ποσοτήτων λιπιδίων στους μύες και, κατά συνέπεια, την παραβίαση της παροχής ενέργειας των τελευταίων. Η καρνιτίνη είναι ένα από τα βασικά συστατικά του συστήματος που παρέχει τη μεταφορά λιπαρά οξέαμε μια μακριά αλυσίδα από το κυτοσόλιο στα μιτοχόνδρια, όπου υφίστανται οξείδωση. Η μυϊκή αδυναμία αναπτύσσεται σε δύο μορφές ανεπάρκειας καρνιτίνης.
Η έλλειψη καρνιτίνης στους μύες αντιπροσωπεύεται κλινικά από προοδευτική αδυναμία των εγγύς ομάδων τους, πιο συχνά σε μαθητές και εφήβους. Μερικές φορές αδυναμία διαλείπουσα και σε συνδυασμό με μυοσφαιρινουρία. Σε σοβαρές περιπτώσεις, μπορεί να εμφανιστεί παράλυση των αναπνευστικών μυών. Τα επίπεδα των ενζύμων στον ορό (κρεατινοκινάση και αλδολάση) είναι αυξημένα. Το ηλεκτρομυογράφημα αποκαλύπτει μη ειδικές αλλαγές χαρακτηριστικές της μυοπάθειας. Στη μυϊκή βιοψία, μπορείτε να δείτε μεγάλο αριθμό σταγονιδίων λίπους. Το επίπεδο της καρνιτίνης στον ορό δεν αλλάζει, αλλά στους μύες μειώνεται. Η αναγνώριση της παθολογίας είναι απαραίτητη, καθώς μπορεί να θεραπευτεί. Συχνά συγχέεται με τη μυϊκή δυστροφία. Το αποτέλεσμα μπορεί να συμβεί μετά από από του στόματος χορήγηση 100 mg / (kg / ημέρα) καρνιτίνης. Σε ορισμένες περιπτώσεις, η θεραπεία με κορτικοστεροειδή είναι αποτελεσματική.
- Η συστηματική ανεπάρκεια καρνιτίνης εκδηλώνεται με προοδευτική μυοπάθεια, συμπεριλαμβανομένης της μυοκαρδιοπάθειας, και ηπατική δυσλειτουργία, συνοδευόμενη από κλινική. ηπατική εγκεφαλοπάθειαΤύπος συνδρόμου Reye. Η ανεπάρκεια καρνιτίνης διαφέρει από την τελευταία ως προς την υποτροπιάζουσα πορεία της και την έντονη μυϊκή αδυναμία που επιμένει μεταξύ των περιόδων έξαρσης της εγκεφαλοπάθειας. Το επίπεδο της κρεατινοφωσφοκινάσης στον ορό είναι σημαντικά αυξημένο, η ποσότητα της καρνιτίνης μειώνεται τόσο στον ορό όσο και στους μύες. Οι αλλαγές στη βιοψία είναι παρόμοιες με εκείνες με ανεπάρκεια καρνιτίνης στον μυϊκό ιστό. Παρόμοιες κλινικές και μορφολογικές αλλαγές, συμπεριλαμβανομένης της ανεπάρκειας καρνιτίνης, μπορεί να ανιχνευθεί σε παραβίαση του μεταβολισμού των οργανικών οξέων, για παράδειγμα, με μεθυλομαλονική και γλουταρική οξέωση (δευτερογενής ανεπάρκεια καρνιτίνης).
Ρύζι. 21-6. Ένα παιδί με συγγενή απουσία του αριστερού μεγάλου μυς του θώρακα.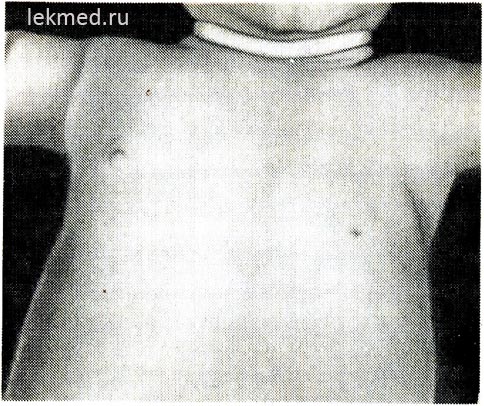
Σημειώστε την απουσία της πρόσθιας μασχαλιαίας πτυχής και της χαμηλής θηλής.
Η θεραπεία συνίσταται στη διατήρηση του ασθενούς σε δίαιτα πλούσια σε υδατάνθρακες και χαμηλά λιπαρά και στη λήψη καρνιτίνης σε ημερήσια δόση 100 mg/kg.
Συγγενείς μυϊκές ανωμαλίες. Συγγενής απουσία μυών. Η μυϊκή υπανάπτυξη μπορεί να είναι αρκετά συχνή και να οδηγήσει σε πλήρη αποκλεισμό των κινήσεων των αρθρώσεων ή σε συγγενή αρθρογρύπωση. Ως εκ γενετής ελάττωμα, ένας μυς λείπει τις περισσότερες φορές. Μια αρκετά συχνή ανωμαλία είναι η απουσία του στέρνου του μείζονος θωρακικού μυός (Εικ. 21-6), σε ορισμένες περιπτώσεις το ελάττωμα αυτό συνδυάζεται με συνδακτυλία στην προσβεβλημένη πλευρά (σύνδρομο Πολωνίας). Η απουσία του θωρακικού μυός συχνά συνοδεύει τη μυϊκή δυστροφία. συγγενής απουσία κοιλιακοι μυςτης κοιλιάς συνδέεται συχνά με ελαττώματα στην ανάπτυξη του ουροποιητικού συστήματος.
Ρύζι. 21-7. Παραμόρφωση αυχένα και ασυμμετρία προσώπου σε αγόρι με συγγενή τορτικόλλη, χωρίς θεραπεία από την ηλικία των 12 ετών. 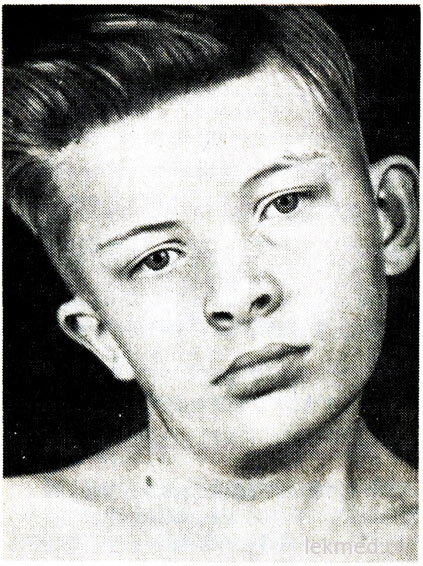
Η συγγενής ταρτικολίτιδα προκαλείται από μονόπλευρη βράχυνση ή σύσπαση του στερνοκλειδομαστοειδούς μυός. Το κεφάλι του ασθενούς έχει κλίση προς τη σύσπαση και το πηγούνι κατευθύνεται προς τα κάτω προς την αντίθετη κατεύθυνση (Εικ. 21-7). Όταν προσπαθείτε να διορθώσετε τη θέση του κεφαλιού, γίνεται αισθητή σημαντική μυϊκή αντίσταση. Στον προσβεβλημένο μυ ψηλαφούνται περιοχές συμπίεσης. Η αιτία του ελαττώματος είναι ασαφής, για μεγάλο χρονικό διάστημα θεωρούνταν το αποτέλεσμα τραυματισμός κατά τη γέννηση. Ωστόσο, η τορτικόλλη εμφανίζεται σε παιδιά που γεννιούνται μέσω χειρουργικής επέμβασης. καισαρική τομή; αυτό υποδηλώνει ότι σε ορισμένες περιπτώσεις η αιτία του ελαττώματος αναφέρεται στην ενδομήτρια περίοδο. Το Torticollis θα πρέπει να διαφοροποιείται από την παθολογική κλίση της κεφαλής λόγω παραμόρφωσης των αυχενικών σπονδύλων, όπως η ανωμαλία Klippel-Weil, και από κατάγματα ή εξαρθρήματα των αυχενικών σπονδύλων. Αποκλείονται με ακτινογραφία. Στα μεγαλύτερα παιδιά, η κλίση της κεφαλής μπορεί να οφείλεται σε στραβισμό, δυστονία, όγκους του οπίσθιου μέρους κρανιακός βόθροςΚαι αυχένιοςνωτιαίος μυελός, οστεοπάθεια μυοσίτιδας, αυχενική λεμφαδενίτιδαή διαφραγματοκήλη. Στις περισσότερες περιπτώσεις συγγενής τορτικολίδαδιορθώνεται με θεραπευτική γυμναστική. Ωστόσο, όταν χρόνια μορφήη τορτικολίς έχει ως αποτέλεσμα μια ασύμμετρη ανάπτυξη του προσώπου και του κεφαλιού (βλ. Εικ. 21-7), η οποία μπορεί να απαιτεί ανατομή του μυός για κοσμητικούς λόγους.
συγγενείς μυοπάθειες. Αυτή η ομάδα περιλαμβάνει αρκετές σπάνιες μορφές κληρονομικών ασθενειών στις οποίες εμφανίζεται μυϊκή αδυναμία και υπόταση ΒΡΕΦΙΚΗ ΗΛΙΚΙΑ(Βλέπε Πίνακα 22-1). Η ακριβής διάγνωσή τους είναι μεγάλης σημασίαςόσον αφορά την πρόβλεψη. Γενικά, είναι ευνοϊκό για τη φυσιολογική δραστηριότητα και το προσδόκιμο ζωής, σε αντίθεση με τη νόσο Werdnig-Hoffmann ή τη συγγενή μυϊκή δυστροφία. Η μυϊκή βιοψία συνήθως βοηθά στον εντοπισμό συγγενών μυοπαθειών.
- Ασθένεια του κεντρικού πυρήνα. Το κεντρικό τμήμα των μυϊκών ινών χρωματίζεται ανώμαλα, αλλά ομοιόμορφα. Μια ηλεκτρονική μικροσκοπική εξέταση αποκαλύπτει μείωση του αριθμού των μιτοχονδρίων και εξάντληση του σαρκοπλασμικού δικτύου στο κεντρικό τμήμα των ινών.
Νεμαλίνη μυοπάθεια. Ο όρος «μη βυσσινί» εξηγείται από το γεγονός ότι οι δομές που μοιάζουν με νήματα προσδιορίζονται στις μυϊκές ίνες.
Ρύζι. 21-8. Μυοτονική σύσπαση της γλώσσας (α) με απότομο χτύπημα με σφυρί κρούσης στο δεξί μισό της και βλέφαρα (β) σε παιδί με υπερκαλιαιμική μορφή οικογενούς περιοδικής παράλυσης. 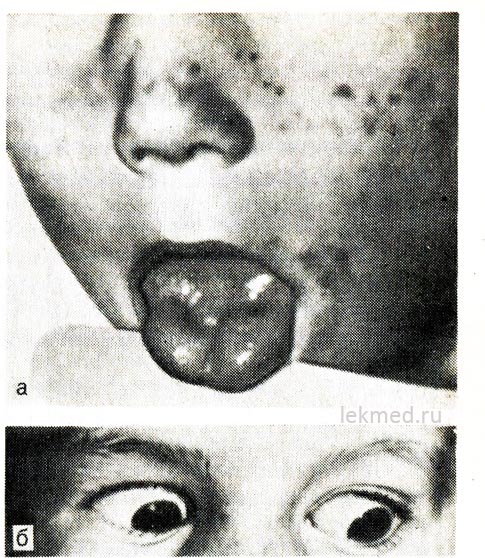
Όταν κοιτάζετε προς τα κάτω, το βλέφαρο παραμένει συσπασμένο.
Τα δεδομένα της ηλεκτρονικής μικροσκοπικής εξέτασης δείχνουν ότι αυτό είναι αποτέλεσμα αλλαγών στις ζώνες Ζ των μυοϊνιδίων.
Μιτοχονδριακές μυοπάθειες. Μερικές μορφές μυοπάθειας έχουν αναφερθεί στις οποίες οι πιο σημαντικές αλλαγές συμβαίνουν στα μιτοχόνδρια των μυϊκών ινών. Μπορούν να αυξηθούν αισθητά τόσο σε αριθμό όσο και σε μέγεθος. Η μυϊκή αδυναμία και η υποτονία μπορούν να προσδιοριστούν ήδη από τη βρεφική ηλικία, αλλά μερικές φορές προοδεύουν αισθητά μόνο στο σχολείο. Η καρδιομυοπάθεια, η εγκεφαλοπάθεια και η γαλακτική οξέωση συχνά συνοδεύουν αυτήν την ομάδα μυοπαθειών.
Μυοτονία. Αυτή η κατάσταση είναι χαρακτηριστικό γνώρισμα διαφόρων μυϊκών ασθενειών, όπως η δυστροφική μυοτονία, η υπερκαλιαιμική οικογενής παροξυσμική παράλυση και οι ασθένειες αποθήκευσης γλυκογόνου. Η μυοτονία ορίζεται ως μια σημαντική καθυστέρηση στη μυϊκή χαλάρωση μετά από εκούσιες ή αναγκαστικές συσπάσεις. Κλινικά εκδηλώνεται με την αδυναμία ξεσφίξεως της γροθιάς ή στην ορατή παρατεταμένη σύσπαση των μυών μετά τη διέγερσή τους, η οποία εκφράζεται με οξύ ερεθισμό (Εικ. 21-8). Αυτό μπορεί να παρατηρηθεί εάν χτυπήσετε μια επιφανειακή ομάδα μυών με ένα σφυρί κρούσης, για παράδειγμα, τους μύες της γλώσσας ή την παλαμιαία επιφάνεια στην περιοχή της ανύψωσης του πρώτου δακτύλου. Η μυοτονία επιβεβαιώνεται με δεδομένα ηλεκτρομυογραφίας. Σε αυτή την περίπτωση, η χαρακτηριστική αυθόρμητη δραστηριότητα των μυών είναι αισθητή μετά τη χαλάρωση ή την εκούσια συστολή τους (μυοτονικές εκκενώσεις).
Συγγενής μυοτονία (νόσος του Τόμσεν). Το μόνο σημάδι αυτής της νόσου, που κληρονομείται από τον κυρίαρχο τύπο, είναι η μυοτονία. Μπορεί να εκδηλωθεί στη βρεφική ηλικία με τη μορφή επιβράδυνσης των κινήσεων κατάποσης και εμετού μετά
επίδραση της αδυναμίας φυσιολογικής χαλάρωσης των μυών του φάρυγγα. Στη μεγαλύτερη παιδική ηλικία, η μυοτονία εκδηλώνεται ως η αδυναμία του ασθενούς να ξεσφίξει τα δάχτυλά του σφιγμένα σε γροθιά. Στην πρώτη προσπάθεια να πραγματοποιήσει κάποιο είδος κίνησης, οι μύες του παιδιού γίνονται σκληροί. Με επαναλαμβανόμενη επανάληψη της ίδιας κίνησης χαλαρώνουν κάπως. Έτσι, για παράδειγμα, ένα άρρωστο παιδί αντιμετωπίζει μεγάλες δυσκολίες στην αρχή της πράξης του περπατήματος. Συνήθως κάνει τα πρώτα βήματα πολύ διστακτικά και αργά. Μετά από λίγα δευτερόλεπτα, το βάδισμα γίνεται φυσιολογικό ή σχεδόν φυσιολογικό. Τα συμπτώματα της μυοτονίας επιδεινώνονται από δυσμενείς συναισθηματική κατάστασηυπομονή και ψύξη του σώματος. Η μυϊκή δύναμη παραμένει φυσιολογική, οι μύες είναι επαρκώς αναπτυγμένοι και συχνά μεγεθύνονται αισθητά, γεγονός που δημιουργεί μια εσφαλμένη εντύπωση για την αθλητική σύσταση του ασθενούς.
Η διάγνωση βασίζεται σε κλινικά ευρήματα και ηλεκτρομυογραφικά δεδομένα. Η ενζυμική δραστηριότητα του ορού είναι εντός φυσιολογικών ορίων. το μοναδικό ιστολογικό σημάδιχρησιμεύει ως υπερτροφία των μυϊκών ινών.
Η ασθένεια διαφέρει από τη δυστροφική μυοτονία λόγω της απουσίας μυϊκής αδυναμίας και ατροφίας και δυστροφικών αλλαγών στη βιοψία του μυϊκού ιστού. Η θεραπεία με νοβοκαΐνη ή θειική κινιδίνη συνοδεύεται από αποτέλεσμα και ενδείκνυται για λειτουργικές διαταραχές. Η πορεία της νόσου είναι συνήθως καλοήθης και η κατάσταση του ασθενούς μπορεί να βελτιωθεί με την ηλικία.
παροξυσμική παράλυση.Αυτή η ομάδα ασθενειών χαρακτηρίζεται από περιοδική μυϊκή αδυναμία με πλήρη ή σχεδόν πλήρη αποκατάσταση της μυϊκής δύναμης στο διάστημα μεταξύ των προσβολών. Περιλαμβάνει επίσης ανεπάρκεια μυϊκής φωσφορυλάσης (νόσος McArdle).
Υπερκαλιαιμική παροξυσμική παράλυση.Η κληρονομική επεισοδιακή αδυναμία, ή παραμυοτονία, μεταδίδεται σύμφωνα με τον κυρίαρχο τύπο και είναι ιδιαίτερα σοβαρή στους άνδρες. Συνήθως ξεκινά από την πρώιμη παιδική ηλικία (μερικές φορές στη βρεφική ηλικία). Οι επιθέσεις συμβαίνουν κατά την περίοδο ανάπαυσης μετά από μεγάλο μυϊκό φορτίο. Η αδυναμία αναπτύσσεται γρήγορα και μπορεί να διαρκέσει αρκετές ώρες. Είναι ιδιαίτερα αισθητό στα πόδια. η αναπνευστική λειτουργία συνήθως δεν επηρεάζεται. Συχνά η αδυναμία συνοδεύεται από μυοτονία, η οποία επιμένει μεταξύ των προσβολών, η οποία εκδηλώνεται πιο ξεκάθαρα με τη μορφή καθυστέρησης στην κίνηση των βλεφάρων κατά το βλέμμα προς τα κάτω (βλ. Εικ. 21-8, β).
Τα επίπεδα καλίου στον ορό είναι συχνά αυξημένα κατά τη διάρκεια μιας επίθεσης, αλλά μπορεί να απαιτούνται πολλαπλές μελέτες κατά τη διάρκεια πολλών προσβολών για να προσδιοριστεί με βεβαιότητα. Είναι δυνατό να προκληθεί τεχνητά μια επίθεση με τη βοήθεια ενός φορτίου καλίου (2-3 g από το στόμα), αλλά θα πρέπει να πραγματοποιείται μόνο υπό έλεγχο ΗΚΓ. Οι επαναλαμβανόμενες επιθέσεις σταματούν από το diacarb. Οι σοβαρές μορφές της νόσου χαρακτηρίζονται από την ανάπτυξη χρόνιας, ήπιας αδυναμίας και εκφυλιστικών αλλαγών στους μύες.
Υποκαλιαιμική παροξυσμική παράλυση. Η οικογενειακή παροξυσμική παράλυση, που επίσης κληρονομείται σύμφωνα με τον κυρίαρχο τύπο, είναι ιδιαίτερα δύσκολη στα αγόρια. Σε αντίθεση με την υπερκαλιαιμική μορφή, η πρώτη προσβολή εμφανίζεται στην ύστερη παιδική ηλικία ή στην πρώιμη παιδική ηλικία. εφηβική ηλικία. Ο λόγος είναι η κατανάλωση ενός πλούσιου γεύματος πλούσιου σε υδατάνθρακες ή η ξεκούραση μετά την άσκηση. Συνήθως η επίθεση ξεκινά το επόμενο πρωί μετά από έντονη σωματική καταπόνηση και ένα πλούσιο δείπνο. Χαρακτηρίζεται από μυϊκή αδυναμία και αρεφλεξία. Η αναπνευστική λειτουργία μπορεί να επηρεαστεί. Μπορεί να ενωθεί αρρυθμία, συμπεριλαμβανομένης της κοιλιακής εξωσυστολίας και της ταχυκαρδίας. Οι επιθέσεις μπορεί να διαρκέσουν περισσότερες από 24 ώρες Στην παραλυτική φάση, το επίπεδο του καλίου στον ορό συνήθως μειώνεται (2-3 mmol / l). Το υποκείμενο ελάττωμα είναι άγνωστο. Οι ασθενείς με επαναλαμβανόμενα σοβαρά επεισόδια αναπτύσσουν χρόνια μυϊκή αδυναμία και παθολογικές αλλαγέςστους μύες. Η θεραπεία κατά τη διάρκεια των επιθέσεων συνίσταται στη λήψη χλωριούχου καλίου. Η αρχική του δόση είναι 2-3 γρ. Το Diakarb βοηθά στη μείωση της συχνότητας των κρίσεων.
Παροξυσμική μυοσφαιρινουρία (ιδιοπαθής μυοσφαιρινουρία). Η ιδιοπαθής μυοσφαιρινουρία είναι μια ετερογενής ομάδα διαταραχών στις οποίες οι κρίσεις παράλυσης με μυοσφαιρινουρία συμβαίνουν αυθόρμητα ή μετά από έντονη άσκηση. Η ασθένεια κληρονομείται με κυρίαρχο τρόπο, συνδεδεμένη με το χρωμόσωμα Χ. Οι μύες, πιο συχνά οι γάμπες και οι μηροί, γίνονται επώδυνοι και πρήζονται κατά τη διάρκεια μιας επίθεσης. Τα ούρα γίνονται σκούρα κόκκινα ή καφέ χρώμα. Η μυοσφαιρινουρία μπορεί να προκαλέσει νεφρική σωληναριακή νέκρωση, η οποία είναι θανατηφόρα λόγω νεφρική ανεπάρκεια.
Η διάγνωση επιβεβαιώνεται με την ανίχνευση μυοσφαιρίνης στα ούρα. Ένα θετικό τεστ βενζιδίνης απουσία ερυθροκυττάρων στα ούρα επιβεβαιώνει την παρουσία μυοσφαιρίνης σε αυτό, ειδικά εάν δεν ανιχνεύεται αιμοσφαιρίνη στον ορό. Η αιμοσφαιρίνη προσδιορίζεται με φασματοφωτομετρία. Η παροξυσμική μυοσφαιρινουρία θα πρέπει να διακρίνεται από τη νόσο McArdle, την ανεπάρκεια της παλμιτυλοτρανσφεράσης της καρνιτίνης και τη μυοσφαιρινουρία μετά από ασυνήθιστη έντονη άσκηση ή μυϊκό τραυματισμό σε υγιές άτομο. Η μυοσφαιρινουρία μετά από έντονη μυϊκή άσκηση εμφανίζεται σε ψευδοϋπερτροφική μυϊκή δυστροφία (νόσος Duchenne).
Η θεραπεία αποτελείται από ανάπαυση στο κρεβάτι. εάν είναι απαραίτητο, πραγματοποιήστε τεχνητό αερισμό των πνευμόνων. Για την πρόληψη της νεφρικής ανεπάρκειας, είναι απαραίτητο να συνταγογραφηθεί άφθονο ποτό στον ασθενή.
Ανεπάρκεια παλμιτυλοτρανσφεράσης καρνιτίνης.Με ανεπάρκεια αυτού του ενζύμου, η μεταφορά λιπαρών οξέων μακράς αλυσίδας στα μιτοχονδριακά τμήματα, στα οποία πραγματοποιείται η οξείδωση και η παραγωγή κετονών, διακόπτεται. Η ανεπάρκεια ισοενζύμου τύπου II κληρονομείται με υπολειπόμενο τρόπο. Λόγω της έλλειψής του, η κετογένεση στους ιστούς, συμπεριλαμβανομένων των μυών και του ήπατος, διαταράσσεται. Τα πρώτα σημάδια της νόσου εμφανίζονται συχνότερα σε παιδιά σχολικής και εφηβικής ηλικίας. Αποτελούνται από επαναλαμβανόμενα επεισόδια μυϊκού πόνου, αδυναμίας και πυρετού μετά από άσκηση ή νηστεία. Η μυοσφαιρινουρία που συνοδεύει τις κρίσεις μπορεί να οδηγήσει σε νεφρική ανεπάρκεια. Η νηστεία οδηγεί σε υπογλυκαιμία. Μεταξύ των επιθέσεων, τα παιδιά φαίνονται υγιή. Η νόσος πρέπει να διαφοροποιείται από άλλες καταστάσεις που συνοδεύονται από περιοδική αδυναμία και μυοσφαιρινουρία. Η μέθοδος για τον προσδιορισμό της δραστικότητας της καρνιτίνης παλμιτυλ τρανσφεράσης έχει διαφορική διαγνωστική αξία. Μειώνεται στους μυϊκούς και ηπατικούς ιστούς, στα λευκοκύτταρα και στην καλλιέργεια ινοβλαστών. Η κατανάλωση μιας διατροφής πλούσιας σε υδατάνθρακες και χαμηλής περιεκτικότητας σε λιπαρά μπορεί να βοηθήσει στη μείωση των επιληπτικών κρίσεων.
Μυϊκές δυστροφίες. Αυτές οι ανωμαλίες ανήκουν σε μια ομάδα οικογενειακών ασθενειών που συνοδεύονται από εκφυλισμό των μυϊκών ινών. Η ταξινόμηση των μυϊκών δυστροφιών βασίζεται σε χαρακτηριστικά όπως ο χρόνος έναρξης, ο ρυθμός εξέλιξης, η κατανομή των βλαβών ανά μυϊκή ομάδα και ο τρόπος κληρονομικότητας.
Ψευδουπερτροφική μυϊκή δυστροφία. Η παιδική ηλικία, ή η μορφή Duchenne, είναι η πιο κοινή μορφή μυϊκής δυστροφίας. η συχνότητά του είναι 0,14 ανά 1000 παιδιά. Στην κλασική μορφή, εμφανίζεται μόνο στα αγόρια και η κληρονομικότητα που συνδέεται με το χρωμόσωμα Χ εμφανίζεται στο 50% περίπου των ανιχνευτών. Σε άλλες περιπτώσεις, η ασθένεια οφείλεται σε νέες μεταλλάξεις. Αναφέρεται μια σπάνια μορφή μυϊκής δυστροφίας, κλινικά πανομοιότυπη με τη μορφή Duchenne, αλλά κληρονομούμενη από έναν υπολειπόμενο τύπο με την ίδια συχνότητα της νόσου σε αγόρια και κορίτσια. Η αξιόπιστη διάγνωση της νόσου είναι σπάνια δυνατή σε ένα παιδί κάτω των 3 ετών. Το ιστορικό συνήθως δείχνει ότι το παιδί είχε καθυστερημένη ανάπτυξη κινητικές λειτουργίες, άρχισε να κάθεται, να περπατάει και να τρέχει αργά, κάτι που φυσικά υποδηλώνει πρώιμη έναρξη της νόσου. Ο βηματισμός (πάπια), η δυσκολία στο ανέβασμα σκαλοπατιών, η υπερτροφία των μυών της γάμπας είναι συχνές. κλινικές ΕΚΔΗΛΩΣΕΙΣ. Σε ορισμένες περιπτώσεις, άλλοι μύες εμπλέκονται επίσης στη διαδικασία, ιδιαίτερα οι μύες του δελτοειδή, του βραχιοειδούς και της γλώσσας.
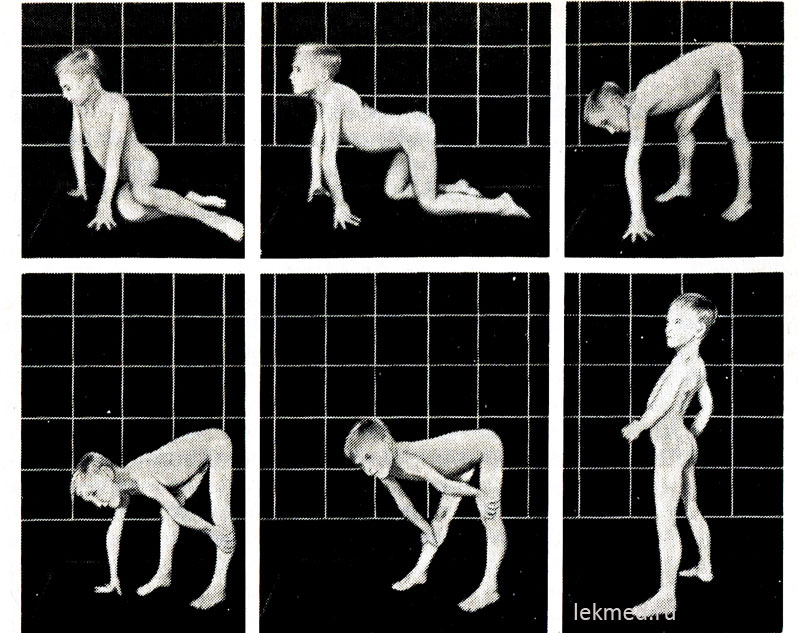
Ρύζι. 21-9. Τυπικές στάσεις που λαμβάνονται όταν σηκώνεται από το πάτωμα (σημάδι Κυβερνήτη) σε ένα 7χρονο παιδί με ψευδουπερτροφική μυοπάθεια.
όρθια θέση ( τελευταία φωτογραφία) αξιοσημείωτη σημαντική λόρδωση.
Στην αρχή της νόσου, οι υπερτροφικοί μύες έχουν σημαντική δύναμη, αλλά αργότερα μειώνεται (ψευδουπερτροφία), αφού η αύξηση της μυϊκής μάζας συμβαίνει λόγω της λιπώδους διήθησής τους. Η δύναμη του υπερτροφισμένου γαστροκνήμιου μυός υπερβαίνει σημαντικά τη δύναμη των μυών της πρόσθιας επιφάνειας του ποδιού, γεγονός που εξηγεί τις συχνές συσπάσεις του τένοντα της πτέρνας και το παιδί που περπατά στα δάχτυλα των ποδιών του. Η αδυναμία των μυών της πυελικής ζώνης εκφράζεται με χαρακτηριστικό βάδισμα πάπιας (άρχοντα) και δυσκολίες που βιώνει το παιδί όταν σηκώνεται από καθιστή θέση στο πάτωμα. Όταν αρκετά σοβαρές μορφέςμυϊκή δυστροφία, το παιδί έχει το σύμπτωμα του Govers: σηκώνεται από το πάτωμα, πρώτα από όλα γονατίζει, ακουμπάει στα χέρια του και μετά σηκώνεται, σπρώχνοντας διαδοχικά τα χέρια του από τις κνήμες του, αρθρώσεις γονάτωνκαι τους μηρούς (Εικ. 21-9). Μπορείτε να προσδιορίσετε την αδυναμία των μυών της ωμικής ζώνης κρατώντας το παιδί σε ανυψωμένη θέση από τις μασχάλες. Κανονικά, προσπαθεί να κρατηθεί πιέζοντας τα χέρια του στο σώμα του. με μυϊκή δυστροφία, φαίνεται να γλιστράει από τα χέρια του εξεταστή. Ένα άρρωστο παιδί συχνά δεν μπορεί να σηκώσει τα χέρια του πάνω από το κεφάλι του. ΣΕ όψιμα στάδιαασθένεια αναπτύσσει σημαντική μυϊκή ατροφία. Συνήθως, μέχρι την ηλικία των 12 ετών, ένα παιδί δεν μπορεί πλέον να περπατήσει. Οι ασθενείς στο 75% των περιπτώσεων πεθαίνουν πριν από την ηλικία των 20 ετών. Οι περισσότεροι από αυτούς έχουν μυοκαρδιοπάθεια, η οποία σε ορισμένες περιπτώσεις προκαλεί αιφνίδιο θάνατο. Εάν η κληρονομικότητα συνδέεται με Χ και η ασθένεια ξεκίνησε σε μεγαλύτερη παιδική ηλικία, το προσδόκιμο ζωής παραμένει μεγάλο (μυϊκή δυστροφία Becker). Μέσος συντελεστής νοητική ανάπτυξησε παιδιά με τη μορφή Duchenne είναι 80? Το 25% των παιδιών έχουν νοητική υστέρηση.
Στο διαφορική διάγνωσηΗ μυϊκή δυστροφία Duchenne θα πρέπει να περιλαμβάνει τη νόσο Werdnig-Hoffmann σε μεγαλύτερα βρέφη και μυϊκές παθήσεις όπως ενδοκρινικές μυοπάθειες, ανεπάρκεια καρνιτίνης, ασθένειες αποθήκευσης γλυκογόνου και πολυμυοσίτιδα. Μερικές φορές με συσπάσεις του τένοντα της πτέρνας και το παιδί που περπατά στα δάχτυλα των ποδιών, μπορεί να υποτεθεί εγκεφαλική παράλυση, αλλά με τη μυϊκή δυστροφία δεν υπάρχουν χαρακτηριστικά σημεία εγκεφαλική παράλυσησπαστικότητα και υπεραντανακλαστικότητα.
Η διάγνωση βασίζεται στον προσδιορισμό της ενζυμικής δραστηριότητας του ορού, των δεδομένων ηλεκτρομυογραφίας και της βιοψίας μυϊκού ιστού. Η δραστηριότητα των ενζύμων, ιδιαίτερα της κρεατινοφωσφοκινάσης, ακόμη και πριν από την ανάπτυξη κλινικά συμπτώματασυχνά υπερβαίνει τον κανόνα κατά 10 φορές ακόμη και στα βρέφη. Στο ηλεκτρομυογράφημα, πρώτα απ 'όλα, αποκαλύπτεται μείωση της διάρκειας και μείωση του εύρους των κινητικών δυναμικών. Οι ιστολογικές αλλαγές συνίστανται σε εκφύλιση των μυϊκών ινών. Συχνά ποικίλλουν σε μέγεθος και εν μέρει αντικαθίστανται από λίπος και συνδετικό ιστό. Το μέγεθος των πυρήνων τους ποικίλλει επίσης. Η διάγνωση μπορεί να τεθεί κατά τη γέννηση με τον προσδιορισμό της δραστηριότητας της κρεατινοφωσφοκινάσης. Μέθοδοι για τον εντοπισμό των θηλυκών φορέων δεν έχουν ακόμη αναπτυχθεί, παρά το γεγονός ότι το 60-80% αυτών εμφανίζει μια ελαφρά ή μέτρια αύξηση στο επίπεδό του. Αυτά τα σημάδια είναι πιο χαρακτηριστικά για την παιδική ηλικία παρά για τις επόμενες περιόδους της ζωής.
Δεν υπάρχουν αποτελεσματικές θεραπείες. Ο ασθενής πρέπει να διατηρείται ενεργός και να μπορεί να περπατά όσο το δυνατόν περισσότερο. Είναι απαραίτητο να διασφαλιστεί ότι το παιδί αποφεύγει την έντονη σωματική δραστηριότητα, καθώς μπορεί να προκαλέσει ρήξη μυϊκών ινών. Σε ορισμένες περιπτώσεις, η χειρουργική επιμήκυνση του τένοντα της πτέρνας βελτιώνει την ικανότητα βάδισης, αλλά παρατεταμένη ξεκούραση στο κρεβάτιμετά από ορθοπεδική διόρθωση μπορεί να αυξήσει τη μυϊκή ατροφία. Η γενετική συμβουλευτική παίζει σημαντικό ρόλο.
Συγγενής μυϊκή δυστροφία. Η νόσος κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο και χαρακτηρίζεται από μυϊκή υπόταση και αδυναμία στο βρέφος. Περιλαμβάνεται στην ομάδα συνθηκών που ορίζονται ως «νωθρό παιδί» (βλ. Πίνακα 21-1). Η εμφάνιση της νόσου αναφέρεται στην ενδομήτρια περίοδο. Μερικές φορές ένα νεογέννητο έχει έντονη ατροφία των μυών, τις συσπάσεις τους, περιορισμένη κινητικότητα των αρθρώσεων. Η διαφοροποίηση από τη νόσο Werdnig-Hoffmann είναι δύσκολη. Οι δεσμίδες της γλώσσας, χαρακτηριστικές της τελευταίας, απουσιάζουν στη μυϊκή δυστροφία. Τα τενοντιακά αντανακλαστικά καταπιέζονται, αλλά δεν χάνονται εντελώς. Η διαδικασία περιλαμβάνει τους μύες που εμπλέκονται στην αναπνοή, συμπεριλαμβανομένου του διαφράγματος. Σε σοβαρές περιπτώσεις, ο θάνατος επέρχεται πριν από την ηλικία του 1 έτους λόγω αναπνευστικής ανεπάρκειας. σε πιο ήπιες μορφές, η φυσιολογική βιωσιμότητα διατηρείται για μεγάλο χρονικό διάστημα. Δεν σημειώνεται αύξηση στη δραστηριότητα των ενζύμων του ορού, αν και εμφανίζονται δυστροφικές αλλαγές στους μύες.
Μορφή μυϊκής δυστροφίας ώμου-προσώπου. Αυτό είναι αρκετό ήπιας μορφήςη μυϊκή δυστροφία κληρονομείται με αυτοσωματικό επικρατή τρόπο. Συνήθως ξεκινά στην ηλικία των 10-20 ετών και χαρακτηρίζεται από αδυναμία και ατροφία των μυών του προσώπου και της ωμικής ζώνης. Το πρόσωπο είναι εντελώς μιμικό, ο ασθενής δεν μπορεί να κλείσει τα μάτια του και να βγάλει σφύριγμα. Η ασθένεια εξελίσσεται αργά και είναι συμβατή με κανονική διάρκειαΖΩΗ. Η διάγνωση βασίζεται στα κλινικά ευρήματα και τον τύπο της κληρονομικότητας. Τα αποτελέσματα μιας βιοψίας μυϊκού ιστού υποδεικνύουν δυστροφικές αλλαγές σε αυτόν. Τα επίπεδα κρεατινοφωσφοκινάσης ορού μπορεί να παραμείνουν φυσιολογικά ή ελαφρώς αυξημένα.
Πυελική μορφή μυϊκής δυστροφίας. Αυτή η ομάδα ετερογενών διαταραχών χαρακτηρίζεται από αργή εξέλιξη της μυϊκής δυστροφίας και κληρονομείται με αυτοσωματικό υπολειπόμενο τρόπο. Η εμφάνιση της νόσου αναφέρεται στη μεγαλύτερη παιδική ηλικία, την εφηβεία ή την ενήλικη ζωή. Συνήθως προσβάλλονται οι μύες της πυελικής ζώνης.
σχήμα ματιούμυοπάθεια. Οι δυστροφικές αλλαγές συμβαίνουν κυρίως στο εξωτερικό μύες των ματιών. Η ασθένεια ξεκινά στην παιδική ή εφηβική ηλικία. Με αυτήν προχωρά η πτώση και ο περιορισμός των κινήσεων. βολβοί των ματιών. Μερικές φορές η αδυναμία επεκτείνεται στους μύες του προσώπου και του λαιμού. Η νόσος πρέπει να διαφοροποιείται από τη μυασθένεια gravis και την παράλυση κρανιακά νεύραμε όγκους του εγκεφαλικού στελέχους.
Η προοδευτική οφθαλμοπληγία, που ξεκινά από την παιδική ηλικία ή την εφηβεία, σχετίζεται με την άτυπη μελαγχρωστική αμφιβληστροειδίτιδα και τον καρδιακό αποκλεισμό (σύνδρομο Kearns-Sayers). Συνήθως σχετίζεται με προοδευτική αταξία, καθυστερημένη ανάπτυξη και εφηβεία. Κάτω από το σαρκόλημμα των μυών, προσδιορίζονται μεγάλες συσσωρεύσεις άτυπων μιτοχονδρίων. Η γενετική φύση αυτής της διαδικασίας δεν έχει τεκμηριωθεί. Μπορείτε να ελέγξετε την πιθανότητα αιφνίδιου θανάτου από παραβίαση της καρδιακής αγωγιμότητας με βηματοδότη.
Μυοτονική δυστροφία. Παρά το γεγονός ότι η μυοτονική δυστροφία ξεκινά σαν σε ενήλικα, η εμφάνισή της καταγράφεται όλο και περισσότερο σε βρέφη και αργότερα παιδιά. Κληρονομείται με αυτοσωμικό κυρίαρχο τρόπο. Η εμφάνισή του στην παιδική ηλικία δείχνει ότι η μητέρα πάσχει από μυοτονία. Σύμφωνα με αυτό, οι ενδομήτριοι παράγοντες μπορούν να επηρεάσουν τη σοβαρότητα της νόσου σε ένα παιδί. Ήδη τη στιγμή της γέννησης μπορεί να έχει μυϊκή υπόταση, του λείπει η ικανότητα να πιπιλίζει. Η υστέρηση της σωματικής και πνευματικής ανάπτυξης συνήθως εντοπίζεται αργότερα. Στην πρώιμη παιδική ηλικία, η μυϊκή αδυναμία και η ατροφία εξαπλώνονται κυρίως στους μύες του προσώπου, της γνάθου και των κροταφικών μυών. Συνήθως σημειώνεται αμφοτερόπλευρη πτώση. Κ διαγνωστικά ουσιαστικές μεθόδουςπεριλαμβάνουν μυϊκή κρούση, ηλεκτρομυογραφία. Χαρακτηριστική για αυτούς τους ασθενείς είναι η αδυναμία να ξεσφίξουν το χέρι σφιγμένο σε γροθιά (βλ. Συγγενής μυοτονία). Η αδυναμία και η ατροφία των μυών των άκρων και της πυελικής ζώνης (συνήθως άπω ομάδες) ανιχνεύονται σε μεγαλύτερη παιδική ηλικία ή εφηβεία. Στους ενήλικες, αυτή η ασθένεια συνοδεύεται από καταρράκτη, φαλάκρα, ατροφία των όρχεων.
Η διάγνωση βασίζεται στον εντοπισμό σημείων μυοτονίας, στη χαρακτηριστική κατανομή της μυϊκής αδυναμίας, στην κληρονομικότητα ανάλογα με τον κυρίαρχο τύπο και στις δυστροφικές αλλαγές στους μύες. Στην παιδική ηλικία, η πορεία της νόσου μπορεί να είναι δυσμενής, συχνά συνοδεύεται από νοητική υστέρηση. ΠΡΟΣ ΤΗΝ εφηβική ηλικίαη μυϊκή αδυναμία έρχεται στο προσκήνιο. Με λειτουργικές διαταραχές, ενδείκνυται θεραπεία με νοβοκαΐνη και κινιδίνη.
Η φλεγμονή είναι μια πολύ επώδυνη ασθένεια που χαρακτηρίζεται από φλεγμονώδεις διεργασίες σκελετικοί μύες, Με ποικίλοι λόγοιπεριστατικό. ΣΕ ιατρική πρακτικήονομάζεται μυοσίτιδα. Το κύριο χαρακτηριστικό είναι γενική αδυναμίαμυών, η οποία προκαλείται από φλεγμονώδεις διεργασίες σε μυϊκούς ιστούςυπεύθυνος για το φορτίο του κινητήρα. Αυτή η φλεγμονή περιλαμβάνει άλλους τύπους που σχετίζονται με λοιμώξεις, τοξίνες και συχνά με επιβλαβείς επιπτώσεις. φάρμακα. Οι κύριες φλεγμονώδεις διεργασίες είναι πολλαπλές και σχετίζονται με επιφανειακή βλάβη.
Ποιος είναι πιο πιθανό να πάθει μυοσίτιδα;
Σύμφωνα με μακροχρόνιες παρατηρήσεις, πιστεύεται ότι η συχνότητα εμφάνισης μιας τέτοιας ασθένειας είναι από δύο έως δέκα περιπτώσεις ανά ένα εκατομμύριο του μέσου ανθρώπινου πληθυσμού. Η ασθένεια χαρακτηρίζεται από δύο ηλικιακές κορυφές εμφάνισης. Η πρώτη εμφανίζεται μεταξύ πέντε και δεκαέξι ετών. Η δεύτερη κορυφή πέφτει σε περισσότερο ανώτερη ομάδασαράντα με εξήντα χρονών. Αποκαλύφθηκε επίσης η προτεραιότητα του φύλου στη νόσο, οι γυναίκες νοσούν περισσότερο από αυτή τη νόσο, η αναλογία άρρωστων γυναικών προς άρρωστους άντρες ποικίλλει, κατά μέσο όρο, από δύο έως τρεις στις γυναίκες σε έναν στους άνδρες.
Ποια είναι τα αίτια της φλεγμονής;
Μέχρι τώρα, τα ακριβή αίτια της εμφάνισης του αυτή η ασθένεια. Όμως είναι η εποχική επίδραση, όσον αφορά τη συχνότητα εμφάνισης, που δεν υποδηλώνει άμεσα αύξηση του αριθμού των ασθενειών σε χειμερινή περίοδοή στις αρχές της άνοιξης, και αυτή είναι ακριβώς η περίοδος ακμής της υπέρβασης των ορίων επιδημίας μεταδοτικές ασθένειες. Αν αναλογιστούμε τη γενετική επίδραση στην εμφάνιση αυτών των φλεγμονών, τότε εμφανίζονται κυρίως σε πανομοιότυπα δίδυμα και συγγενείς σε ευθεία γραμμή που είχαν ήδη μια τέτοια ασθένεια. Αυτή η μεταφορά γονιδιακών πληροφοριών συνήθως δεν σχετίζεται με την ίδια την ασθένεια, αλλά σχετίζεται με συγκεκριμένες διαταραχές στο ανοσοποιητικό σύστημα.
Τα πρώτα σημάδια μυοσίτιδας
Οι κύριοι δείκτες της πορείας της νόσου είναι οι διαταραχές στις ανοσολογικές αποκρίσεις κυτταρικό επίπεδο. Μια ανοσοπαθολογική εξέταση της φλεγμονώδους περιοχής των μυών αποκαλύπτει παθολογική διήθηση ορισμένων κυτταρικών σωμάτων που βρίσκονται στην αρχική ενεργή κατάσταση και απελευθερώνει μια τοξίνη που επηρεάζει τις διαδικασίες αποκατάστασης. Με τη δερματική εκδήλωση της νόσου, είναι δυνατό να εντοπιστούν ορισμένα παθογόνα που παραβιάζουν τη συνολική κυτταρική αντίσταση. Ανιχνεύεται επίσης μια αντίδραση αντισωμάτων, η οποία σε ορισμένες περιπτώσεις έχει αρνητικές αντιδράσεις στα αγγεία.
Στην αρχική πορεία της νόσου γίνονται συχνές επισκέψεις ασθενών λόγω αδιαθεσίας και μεγάλης αδυναμίας και φλεγμονωδών διεργασιών στο δέρμα. Με την πάροδο του χρόνου, αρκετές εβδομάδες, παρατηρείται αύξηση των αρχικών ενδείξεων και αύξηση της αδυναμίας και, κινητικοί μύεςαχ χέρια και πόδια. Σε περίπτωση ασθένειας νεαρού οργανισμού, παρατηρούνται οξεία αρχικά συμπτώματα, που συνοδεύονται από έντονες αλλαγές στη σύσταση του σώματος, είτε απώλεια βάρους, είτε εμπύρετα φαινόμενα.
Αν λάβουμε υπόψη τους ηλικιωμένους ασθενείς, τότε παρατηρούν σταδιακή αύξηση της αδυναμίας στους μύες, μερικές φορές σε αρκετά χρόνια. Σπάνια, επιφανειακές βλάβες δέρμα. Μερικοί ασθενείς μπορεί επίσης να εμφανίσουν συγκεκριμένα σημάδια, που συνοδεύονται από σπασμούς αιμοφόρων αγγείων και κυκλοφορικές διαταραχές, διαταραχές σε οστικό ιστόκαι δυσκολία στην αναπνοή, η οποία εμφανίζεται λόγω ορισμένων φλεγμονωδών διεργασιών στους πνεύμονες.
Πώς εκδηλώνεται η ασθένεια;
Η πρώτη εκδήλωση αυτής της ασθένειας είναι κλινικό σημείοΗ όραση είναι μια γενική αδυναμία όλων των μυϊκών ομάδων που είναι υπεύθυνες για την κινητική δραστηριότητα και του άνω και κατώτερες ομάδεςαυτούς τους μύες, καθώς και την περιοχή του τραχήλου της μήτρας. Αυτές οι διαταραχές οδηγούν στο γεγονός ότι ο ασθενής κινείται πολύ σκληρά στην αρχική φάση μετά από μεγάλη ανάπαυση, είναι δύσκολο να σηκωθεί από καθιστή θέση ή να μπει ή να βγει από τη μεταφορά ή σε άλλες καθημερινές κινήσεις. Από την πλευρά, οι διαταραχές στο βάδισμα είναι ορατές, δεν είναι ξεκάθαρο, δεν είναι πολύ συντονισμένο.
Υπάρχουν περιπτώσεις που ο ασθενής δεν μπορεί να σηκωθεί μόνος του από το κρεβάτι. Η φλεγμονή των μυών μπορεί επίσης να επηρεάσει την περιοχή που σχετίζεται με την κατάποση και τη φωνή, αυτό μπορεί να συνοδεύεται από οξεία δυσκολία στις προσπάθειες κατάποσης, που καταλήγει σε εκρηκτικό βήχα. Επίσης, ορισμένοι ασθενείς αναπτύσσουν δερματομυοσίτιδα, η οποία έχει εκδηλώσεις με τη μορφή περιοχών με αυξημένο εξάνθημα σε ορισμένα σημεία του σώματος ή στο πρόσωπο ή το λαιμό και σε άλλα μέρη του σώματος. Υπάρχουν και άλλοι δερματικές εκδηλώσεις, που υποδηλώνουν άμεσα την πορεία της νόσου, μπορούν επίσης να συνδυαστούν με εκδηλώσεις πολυμυοσίτιδας, μπορεί να είναι φολιδωτές και ερυθρές περιοχές του δέρματος ή ακόμη και να συνοδεύονται από μικρορωγμές του δέρματος σε ορισμένες περιοχές των άνω άκρων.
Εναπόθεση αλάτων ασβεστίου στους μαλακούς ιστούς
Η εναπόθεση αλάτων στους μαλακούς ιστούς (ασβεστοποίηση) είναι συνήθως προϊόν μιας μακροχρόνιας ασθένειας, όψιμα στάδια φλεγμονώδεις διεργασίες. Οι σχηματισμοί εμφανίζονται και εντοπίζονται συνήθως κάτω από το δέρμα ή σε σημεία ιστών που συνδέουν τους μύες, πάνω από τα αρθρικά μέρη των γονάτων και των αγκώνων, πάνω από τις φάλαγγες των δακτύλων και πολλά άλλα, ακόμη και σε ορισμένες περιοχές των γλουτών.
Φλεγμονώδεις διεργασίες στους πνεύμονες με μυοσίτιδα
Ένα σαφές αρχικό σημάδι φλεγμονωδών διεργασιών στους πνεύμονες με μυοσίτιδα είναι η αναπνευστική δύσπνοια, συνήθως σχετίζεται με παραβίαση των μυών του διαφράγματος ή λάθος δουλειάκαρδιά ή με φλεγμονή που σχετίζεται με τοξίνες που περιέχονται στα φάρμακα που λαμβάνει ο ασθενής. Επίσης, οι φλεγμονώδεις διεργασίες συνοδεύονται από βήχα ή ανεπαρκή αναπνευστική δραστηριότητα. Συγκεκριμένα δύσκολες περιπτώσειςμπορεί να αναπτυχθεί ασφυκτική πνευμονία.
Πώς επηρεάζει η μυοσίτιδα την καρδιά;
Οι φλεγμονώδεις διεργασίες που συμβαίνουν στον καρδιακό μυ με μυοσίτιδα συνήθως προχωρούν χωρίς έντονα συμπτώματα. Μόνο όταν ειδική μελέτημε τη βοήθεια του καρδιογραφήματος παρατηρούνται κάποιες αστοχίες στους ρυθμούς του παλμού που συνήθως αποδίδονται σε περιοχές άλλων ασθενειών. Καρδιακή ανεπάρκεια πρακτικά δεν παρατηρείται.
Διάγνωση φλεγμονής σε εργαστηριακές μελέτες
Κατά τη διεξαγωγή αιματολογικών εξετάσεων, δεν παρατηρούνται σοβαρές αλλαγές. Άλλες αναλύσεις αποκαλύπτουν αποκλίσεις που είναι χαρακτηριστικές διαταραχών των μυών που είναι υπεύθυνοι για την κινητική δραστηριότητα (CPK). Σε ορισμένες πορείες της νόσου, παρατηρούνται σε όλους σχεδόν τους ασθενείς. Ο κορεσμός αυτού του δείκτη μπορεί να αυξηθεί έως ότου εντοπιστούν ξεκάθαρα αρχικά σημάδια κατά τη διάρκεια κλινική εξέταση. Αλλά μερικές φορές αυτός ο δείκτης μπορεί να είναι εντός του φυσιολογικού εύρους και να μην υποδηλώνει καν σοβαρή φλεγμονή του μυϊκού ιστού. Υπάρχει επίσης μια ένδειξη με χαμηλή ικανότητα κινητήρα που υποδεικνύει κάποια πιθανή γενικά συμπτώματαμε ηπατίτιδα (τρανσμινάσες).
Θεραπεία μυϊκής φλεγμονής
αποκαλύφθηκε στην πράξη. Ότι η μυϊκή φλεγμονή είναι μια ασθένεια που προχωρά σύμφωνα με ένα περιοδικό μοτίβο, δηλαδή με παροξύνσεις. Πόνος που εμφανίζεται με άλλους χρόνιες ασθένειεςσταθερή, και στην περίπτωση αυτής της ασθένειας, περιοδική, συνεχώς μην ενοχλείτε. Είναι η μυοσίτιδα που έχει δύο στάδια της πορείας της νόσου - χρόνια και οξεία. Με την κατάλληλη θεραπεία, το οξύ στάδιο της νόσου μετατρέπεται ομαλά σε χρόνιο, όχι τόσο επώδυνο. Κατά τη διάρκεια του χρόνιου σταδίου της νόσου, οι φλεγμονώδεις διεργασίες γίνονται ευαίσθητες σε αλλαγές της εποχής ή σε κλιματική αλλαγή. χρόνια φλεγμονήμπορεί να προκληθεί όχι μόνο ως στάδιο μετά την οξεία φάση της μυοσίτιδας. Αλλά και με κάποιες ασθένειες μολυσματικού χαρακτήρα. Ως εκ τούτου, συχνά, οι άνθρωποι δεν γνωρίζουν καν τη μυοσίτιδα όταν είναι άρρωστοι με τη συνήθη μολυσματική γρίπη, αλλά μετά την ανάρρωση εμφανίζονται σημάδια μυϊκής βλάβης. Οξεία φλεγμονήοι μύες μπορούν να εκδηλωθούν σε μια τοπική τοποθεσία.
Είναι πολύ δύσκολο να εντοπιστεί η ασθένεια μυοσίτιδα, επειδή είναι αυστηρά περιοδική. Αλλά με τα πρώτα συμπτώματα, πρέπει να επικοινωνήσετε με έναν ειδικό. Αυτό πρέπει να γίνει για διάφορους λόγους, αλλά ο πρώτος πρέπει να είναι ο ορισμός μιας ακριβούς διάγνωσης και μετά το ραντεβού βέλτιστη θεραπεία, το κυριότερο είναι ότι θα είχε συστημικό χαρακτήρα.
Η θεραπεία της μυϊκής φλεγμονής μπορεί να επιφέρει τη σωστή κατανομή του φόρτου εργασίας στο σώμα με σωματική επίδραση στη νόσο. Σε ορισμένες περιπτώσεις χρησιμοποιούνται αθλητικά φορτία που εναλλάσσονται με περιόδους πλήρους χαλάρωσης. Η πρώτη προτεραιότητα στη θεραπεία της μυοσίτιδας είναι να ανακαλύψουμε τα αίτια της φλεγμονής. Με βάση την έρευνα, φαρμακευτική θεραπείαμε τη βοήθεια εξειδικευμένων φαρμάκων. Συνήθως, ένα σύμπλεγμα φαρμάκων συνταγογραφείται για θεραπεία, αλλά σε κάθε σύστημα περιλαμβάνονται παυσίπονα που βασίζονται σε αναλγητικά και φάρμακα που αποτρέπουν τις φλεγμονώδεις διεργασίες. Χρησιμοποιούνται πολλά φάρμακα, αλλά τα κυριότερα είναι τα Diclofenac, Ketonal, Nurofen.
Με τοπικές ή τοπικές εστίες της νόσου, χρησιμοποιούνται ειδικά σκευάσματα, τα οποία συνταγογραφούνται με τη μορφή αλοιφών θερμαντικού τύπου: Apizatron, Nikoflex, Finalgon. Στη θεραπεία των φλεγμονωδών διεργασιών στα παιδιά, συνήθως συνταγογραφούνται εξειδικευμένα σκευάσματα με μειωμένη δόση των θεραπευτικών συστατικών, σε αυτή η υπόθεσηΥπάρχει καλή ανατροφοδότησηγια τη Δρ.
Σε εκείνες τις περιόδους που υπάρχει έξαρση της πορείας της νόσου, σε καμία περίπτωση δεν πρέπει να φορτώνετε το σώμα σωματική δραστηριότηταΚαι γενικά πρέπει να τηρείτε την ανάπαυση στο κρεβάτι. Αυτό είναι υψίστης σημασίας στις φλεγμονώδεις διεργασίες στην περιοχή των κινητικών μυών της σπονδυλικής στήλης. Με μια τέτοια έξαρση, είναι απαραίτητο να ληφθεί τα ακόλουθα φάρμακααναλγητική δράση και φάρμακα που αποτρέπουν τις φλεγμονώδεις διεργασίες, ο κατάλογος τέτοιων φαρμάκων περιλαμβάνει: Brufen, Reopirin, Indomethacin.
Μια τέτοια θεραπεία μπορεί να είναι αποτελεσματική μόνο σε αυτή την περίπτωση. Εάν είναι συστηματικό και χρησιμοποιείται σε συνδυασμό με άλλα αποτελεσματικές μεθόδουςκαι μέσα. Από αυτό προκύπτει ότι εκτός από τη λήψη φαρμάκων, πρέπει να πραγματοποιούνται φυσιοθεραπευτικές διαδικασίες και είναι πολύ επιθυμητό να χρησιμοποιούνται ασκήσεις που περιλαμβάνονται στη θεραπευτική και προληπτική φυσική αγωγή. Αλλά πρέπει να θυμόμαστε ότι στην οξεία μυοσίτιδα των κινητικών μυών της σπονδυλικής στήλης, μια τέτοια φυσική αγωγή αντενδείκνυται αυστηρά. Με αύξηση γενική θερμοκρασίασώμα κατά τη διάρκεια της νόσου, είναι απαραίτητο να χρησιμοποιείτε φάρμακα που το μειώνουν και φροντίστε να περιορίσετε την επαφή με την ψυχρή ατμόσφαιρα.
Σε περιπτώσεις όπου οι φλεγμονώδεις διεργασίες του άνω μέρους των μυών που ευθύνονται για το κινητικό φορτίο στον αυχένα προκαλούνται από σηπτικές βλάβες, τότε μετά από οριστική διαδικασία, είναι απαραίτητο να επικοινωνήσετε με έναν χειρουργό, ο οποίος είναι υποχρεωμένος να ανοίξει αυτή τη ζώνη στο έναν λειτουργικό τρόπο και αφαιρέστε ολόκληρη τη μόλυνση. Επίσης, με αυτό το είδος μυϊκής φλεγμονής, κάθε είδους μασάζ απαγορεύεται αυστηρά.
Κάθε τύπος μυοσίτιδας έχει τα δικά του, χαρακτηριστικά μόνο για αυτόν, χαρακτηριστικά. Για παράδειγμα, μια φλεγμονώδης διαδικασία στην αυχενική περιοχή μιας μυϊκής ομάδας αντιμετωπίζεται χωρίς ιδιαίτερες επιπλοκές και είναι σχετικά εύκολη εάν αυτή η θεραπεία δεν ξεκίνησε σε προχωρημένη νόσο, αλλά αμέσως με την εμφάνιση των πρώτων σημείων. Στη θεραπεία αυτού του τύπου μυοσίτιδας, οι ειδικοί συνήθως συνταγογραφούν καθιστική θεραπεία ή, το καλύτερο από όλα, ανάπαυση στο κρεβάτι. Οι θεράποντες ειδικοί συνταγογραφούν ένα σύμπλεγμα φαρμάκων, συμπεριλαμβανομένων των θερμαντικών αλοιφών, τα οποία τρίβονται στην περιοχή της φλεγμονής και λαμβάνουν φάρμακα που αποτρέπουν τις φλεγμονώδεις διεργασίες.
Στην πράξη, το λεγόμενο «μπλοκάρισμα της νοβοκαΐνης» δίνει ένα εξαιρετικό αποτέλεσμα. Η διαδικασία αυτής της εκδήλωσης συνίσταται στην εφαρμογή ενός αναισθητικού γύρω από τη φλεγμονώδη περιοχή με Novocain και ειδική ορμόνη. Αλλά δίνει θετικό αποτέλεσμαμόνο εάν ο ασθενής δεν έχει αλλεργικές αντιδράσειςκαι αντενδείξεις. Υπάρχει επίσης μια μέθοδος διάτασης των μυών και των συνδέσμων για μια διαδικασία χαλάρωσης. Είναι απολύτως νέα μέθοδος, που δίνει εξαιρετικά αποτελέσματα και έχει ήδη δοκιμαστεί σε πολλά ιατρικά ιδρύματα.
Στο συνημμένο βίντεο, μπορείτε να ανακαλύψετε γιατί οι τένοντες έχουν φλεγμονή.
Αλλά οι πιο απλές συστάσεις που συνοδεύουν κάθε υποψήφιο ασθενή καλός ειδικός, είναι συστάσεις για την προστασία των κρίσιμων περιοχών των μυών από το κρύο, για να μην λιμνάζουν και να μην μένουν σε μια στατική θέση για πολλή ώρα, τόσο όταν είστε ξαπλωμένοι όσο και όταν κάθεστε. Επιλέξτε μόνο άνετες στάσεις στις οποίες οι μύες δεν μουδιάζουν, αποκλείστε τα ρεύματα και πραγματοποιήστε γενικές ασκήσεις ενδυνάμωσης. Αυτές οι απλές και μάλλον απλές συστάσεις θα σας βοηθήσουν να μην εκτεθείτε άσκοπα από φλεγμονώδεις διεργασίες και μυοσίτιδα.